Dear Prof. Gale,
Thank you for your answer and GULP in general! 
I am sorry for two more questions, but I could not find any information about them anywhere.
I noted the unusual behavior of the default optimization algorithm, which occurred at the parallel version of GULP (compiled via intel-parallel-studio. 2020.4).
While I execute the same input file (constrained molecular crystal relaxation) with a different number of cores, I get different results (energy and geometry).
The difference happens after 3–4 relaxation steps; for instance, here is a small comparison of output executed at 8 and 10 cores:
8 cores:
Cycle: 0 Energy: 220.895317 Gnorm: 0.158248 CPU: 0.531
Hessian calculated
Cycle: 1 Energy: 220.893199 Gnorm: 0.155718 CPU: 0.569
Hessian calculated
Cycle: 2 Energy: 220.807090 Gnorm: 0.173120 CPU: 0.626
Hessian calculated
Cycle: 3 Energy: 220.574387 Gnorm: 0.172520 CPU: 0.680
Hessian calculated
Cycle: 4 Energy: 220.232424 Gnorm: 0.116935 CPU: 0.727
Hessian calculated
Cycle: 5 Energy: 218.856032 Gnorm: 0.152991 CPU: 0.791
10 cores:
Cycle: 0 Energy: 220.895317 Gnorm: 0.158248 CPU: 0.252
Hessian calculated
Cycle: 1 Energy: 220.893199 Gnorm: 0.155718 CPU: 0.287
Hessian calculated
Cycle: 2 Energy: 220.807090 Gnorm: 0.173120 CPU: 0.336
Hessian calculated
Cycle: 3 Energy: 220.246853 Gnorm: 0.169928 CPU: 0.384
Hessian calculated
Cycle: 4 Energy: 220.036451 Gnorm: 0.144967 CPU: 0.422
Hessian calculated
Cycle: 5 Energy: 217.328074 Gnorm: 0.276808 CPU: 0.489
I can’t understand what happened after Cycle 2, because Energy and Gnorm before that step in both cases are the same.
Similar differences happened between serial and parallel implementations, but here it’s logical due to the difference between 2D hessian and lower half triangular.
May I ask for a possible reason for that and what the most reliable setting is for parallel calculations?
Keywords of my calculations:
optimise molmec rigid kcal
stepmx 0.1
My other question regarding rigid relaxation with the same input setting as previously
Very often, when I perform such calculations, it looks like the calculation will converge to a certain energy value, but instantly energy enormously increases and tries to escape from that further.
Here is an example:
Cycle: 137 Energy: 210.087631 Gnorm: 0.003501 CPU: 19.464
Cycle: 138 Energy: 210.087504 Gnorm: 0.003393 CPU: 19.611
Cycle: 139 Energy: 210.087478 Gnorm: 0.003382 CPU: 19.733
Cycle: 140 Energy: 287842468.380507 Gnorm:************** CPU: 20.092
Hessian calculated
Cycle: 141 Energy: 286876014.965150 Gnorm:************** CPU: 20.364
Hessian calculated
…
Hessian calculated
Cycle: 152 Energy: 187110.248387 Gnorm: 507962.052378 CPU: 23.428
Hessian calculated
Cycle: 153 Energy: 3650.966662 Gnorm: 7442.229716 CPU: 23.818
Hessian calculated
Cycle: 154 Energy: 312.351900 Gnorm: 105.817730 CPU: 24.209
Hessian calculated
Cycle: 155 Energy: 247.417067 Gnorm: 19.156643 CPU: 24.425
…
Cycle: 241 Energy: 201.442604 Gnorm: 0.299935 CPU: 38.733
Cycle: 242 Energy: 200.534914 Gnorm: 0.194973 CPU: 38.908
Cycle: 243 Energy: 200.046791 Gnorm: 0.140701 CPU: 39.034
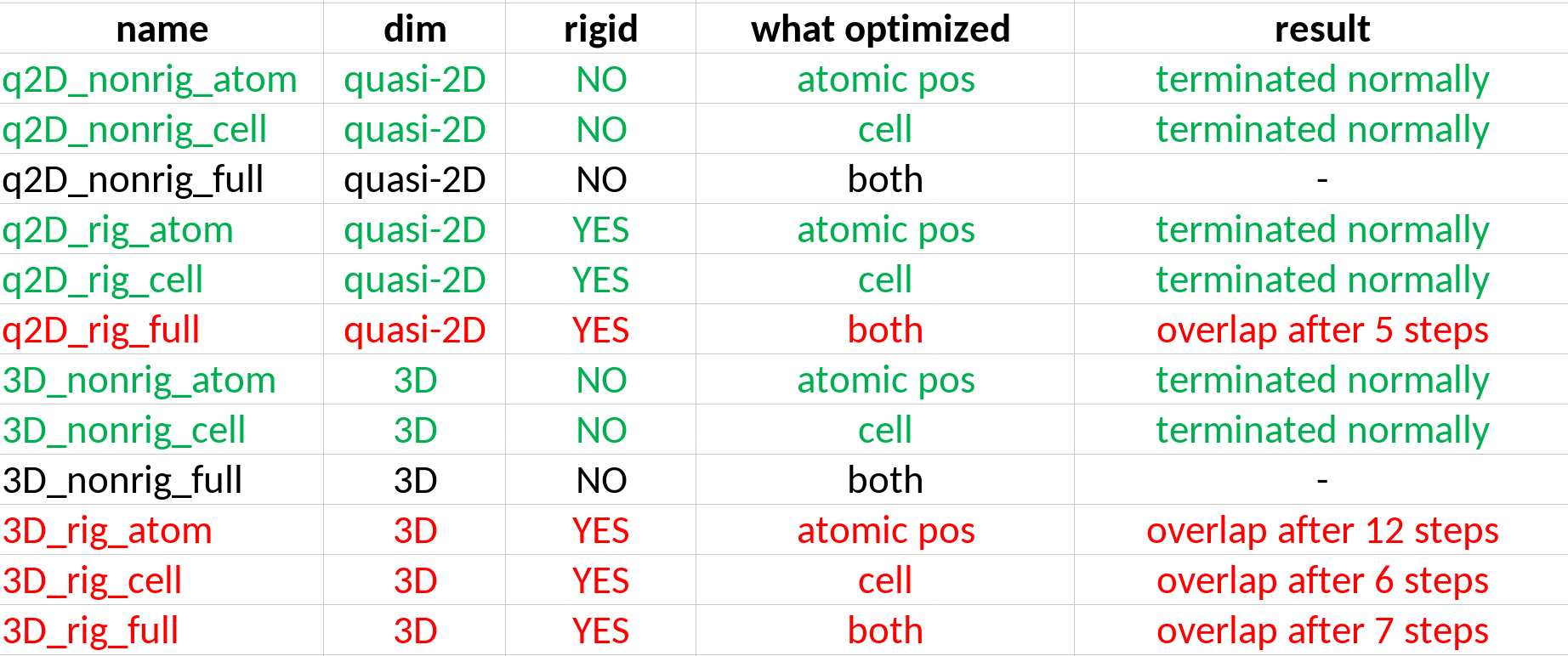
I checked it in with a series of different input crystal geometry configurations but the same compound, and very often optimization gets stuck in such a “high energy” configuration.
Sometimes the method can jump away from it.
In general, it is related to molecules overlapping, but I thought because I chose rigid body relaxation, the program avoided such trapping. Input geometries far away from being overlapped.
Please, can you give some insight on how to avoid it?
Sorry again for such a lot of text.
Thank you in advance!
Best,
Alexey