Hi, lammps-users,
I need to put K+(H2O)6 and Ni2+(H2O)6 in a water box and do some dynamics.
The force field used is UFF, and I fit Ni and K parameter according to literature (Addicoat, M. A.; Vankova, N.; Akter, I. F.; Heine, T., Extension of the universal force field to metal–organic frameworks. Journal of chemical theory and computation 2014, 10 (2), 880-891.).
If put K+(H2O)6 or Ni2+(H2O)6 in a empty box and do minimize, then the structure maintains steady.
If put Ni2+(H2O)6 in a water box and do minimize, the structure maintains steady, too.
But If put K+(H2O)6 in a water box and do minimize, K+(H2O)6 turns distorted after minimization.
I don’t know why, the only thing different here is the bond length of Ni-O(~2.0A) is much short than K-O(~2.7A).
please give me some advice.
trj before minimize(f1 ,f2) and after(f3 ,f4) (set dynamic bond in VMD):
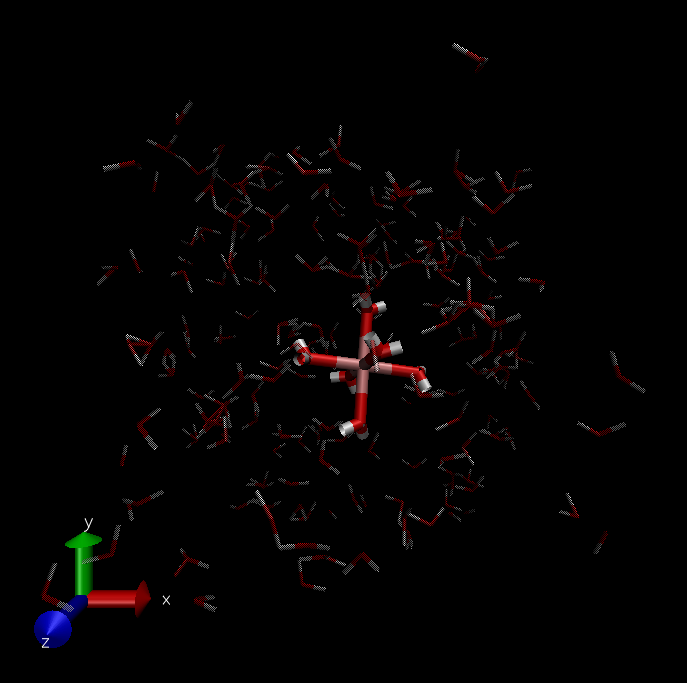
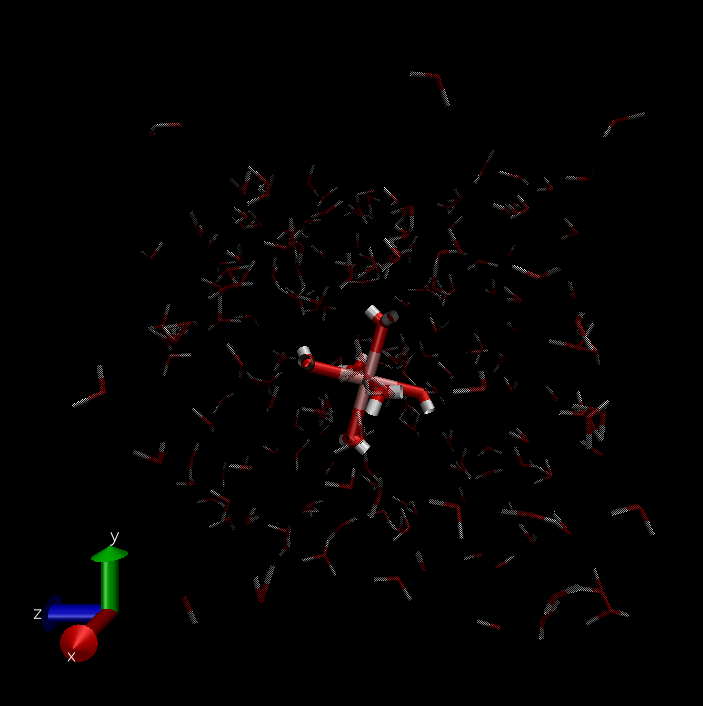


The input script is list below:
#data.K+H2O6
Created on Thu Apr 26 21:52:18 2018
19 atoms
18 bonds
33 angles
3 atom types
2 bond types
3 angle types
0.000000 20.000000 xlo xhi
0.000000 20.000000 ylo yhi
0.000000 20.000000 zlo zhi
0.000000 0.000000 0.000000 xy xz yz
Masses
1 39.098300000 # K_6+1
2 1.007940000 # H_
3 15.999400000 # O_3
Bond Coeffs
1 39.742249 2.818422 # K_6+1 O_3
2 559.995206 0.990254 # H_ O_3
Angle Coeffs
1 cosine/periodic 10.476559 1 4 # O_3 K_6+1 O_3
2 fourier 6.015281 0.300235 0.267331 0.266745 # K_6+1 O_3 H_
3 fourier 122.883342 0.300235 0.267331 0.266745 # H_ O_3 H_
Pair Coeffs
1 0.035000 3.396106 # K_6+1 K_6+1
2 0.044000 2.571134 # H_ H_
3 0.060000 3.118146 # O_3 O_3
Atoms
1 1 1 0.00000 10.66680 -10.90100 -10.06280
2 1 2 0.00000 12.25080 -9.95660 -7.26040
3 1 3 0.00000 11.32120 -10.21140 -7.39940
4 1 2 0.00000 10.82040 -9.47120 -7.01240
5 1 2 0.00000 10.74100 -7.52760 -10.14180
6 1 3 0.00000 10.89560 -8.19740 -10.83160
7 1 2 0.00000 11.73840 -7.92300 -11.23500
8 1 2 0.00000 13.69780 -11.25560 -11.52400
9 1 3 0.00000 13.39480 -11.30020 -10.59960
10 1 2 0.00000 13.80500 -12.11540 -10.26000
11 1 2 0.00000 10.36860 -12.41140 -13.06920
12 1 3 0.00000 9.99360 -11.58360 -12.71900
13 1 2 0.00000 9.04180 -11.66360 -12.90860
14 1 2 0.00000 9.63980 -13.91240 -8.91680
15 1 3 0.00000 10.47560 -13.61740 -9.32040
16 1 2 0.00000 10.63740 -14.27320 -10.02180
17 1 2 0.00000 7.48640 -9.76280 -9.87220
18 1 3 0.00000 7.94520 -10.53160 -9.48880
19 1 2 0.00000 7.64760 -10.54160 -8.56180
Bonds
1 1 1 3
2 1 1 6
3 1 1 9
4 1 1 12
5 1 1 15
6 1 1 18
7 2 2 3
8 2 3 4
9 2 5 6
10 2 6 7
11 2 8 9
12 2 9 10
13 2 11 12
14 2 12 13
15 2 14 15
16 2 15 16
17 2 17 18
18 2 18 19
Angles
1 1 3 1 15
2 1 6 1 9
3 1 18 1 3
4 1 18 1 12
5 1 9 1 15
6 1 18 1 15
7 1 18 1 9
8 1 12 1 15
9 1 18 1 6
10 1 3 1 6
11 1 6 1 12
12 1 3 1 12
13 1 3 1 9
14 1 6 1 15
15 1 9 1 12
16 2 1 3 2
17 3 2 3 4
18 2 1 3 4
19 2 1 6 5
20 3 5 6 7
21 2 1 6 7
22 2 8 9 1
23 2 1 9 10
24 3 8 9 10
25 2 1 12 13
26 2 1 12 11
27 3 11 12 13
28 2 16 15 1
29 3 16 15 14
30 2 1 15 14
31 2 1 18 19
32 2 1 18 17
33 3 19 18 17
#in.K+H2O6
log log.K+H2O6 append
units real
atom_style full
boundary p p p
pair_style lj/cut/coul/long 10.000
bond_style harmonic
angle_style hybrid fourier cosine/periodic
kspace_style ewald 0.000001
dielectric 1.0
pair_modify tail yes mix arithmetic
special_bonds lj/coul 0.0 0.0 1.0
box tilt large
read_data data.K+H2O6 extra/atom/types 2 extra/bond/types 1 extra/angle/types 1
insert H2O
molecule h2o H2O.txt offset 3 2 3 0 0
create_atoms 0 random 200 1 NULL mol h2o 1
mass 4 15.9994 # O
mass 5 1.008 # H
pair_coeff 4 4 0.060000 3.118146
pair_coeff 5 5 0.044000 2.571134
bond_coeff 3 559.995206 0.990254
angle_coeff 4 fourier 122.883342 0.300235 0.267331 0.266745
insert H2O
Atom Groupings
group K id 1:19
END Atom Groupings
QEQ
set type 1 charge 1
fix qeqK K qeq/dynamic 1 10 1.0e-3 200 param.qeq1
run 0
unfix qeqK
QEQ
dump trjmini all xyz 10 mini.xyz
dump_modify trjmini element K H O O H
minimize 1.0e-8 1.0e-8 10000 10000
write_data data.aftermini
undump trjmini
#in.Ni2+H2O6
log log.Ni2+H2O6 append
units real
atom_style full
boundary p p p
pair_style lj/cut/coul/long 10.000
bond_style harmonic
angle_style hybrid cosine/periodic fourier
kspace_style ewald 0.000001
special_bonds lj/coul 0.0 0.0 1.0
dielectric 1.0
pair_modify tail yes mix arithmetic
box tilt large
read_data data.Ni2+H2O6 extra/atom/types 2 extra/bond/types 1 extra/angle/types 1
insert H2O
molecule h2o H2O.txt offset 3 2 3 0 0
create_atoms 0 random 200 1 NULL mol h2o 1
mass 4 15.9994 # O
mass 5 1.008 # H
pair_coeff 4 4 0.060000 3.118146
pair_coeff 5 5 0.044000 2.571134
bond_coeff 3 559.995206 0.990254
angle_coeff 4 fourier 122.883342 0.300235 0.267331 0.266745
insert H2O
Atom Groupings
group Ni id 1:19
END Atom Groupings
QEQ
set type 1 charge 2
fix qeqNi Ni qeq/dynamic 1 10 1.0e-3 200 param.qeq2
run 0
unfix qeqNi
QEQ
dump trjmini all xyz 10 mini.xyz
dump_modify trjmini element K H O O H
minimize 1.0e-8 1.0e-8 10000 10000
write_data data.aftermini
undump trjmini
#data.Ni2+H2O6
Created on Thu Apr 26 21:29:29 2018
19 atoms
18 bonds
33 angles
3 atom types
2 bond types
3 angle types
0.000000 20.000000 xlo xhi
0.000000 20.000000 ylo yhi
0.000000 20.000000 zlo zhi
0.000000 0.000000 0.000000 xy xz yz
Masses
1 58.693400000 # Ni6+2
2 1.007940000 # H_
3 15.999400000 # O_3
Bond Coeffs
1 165.282024 2.239280 # Ni6+2 O_3
2 559.995206 0.990254 # H_ O_3
Angle Coeffs
1 cosine/periodic 33.090760 1 4 # O_3 Ni6+2 O_3
2 fourier 30.853750 0.300235 0.267331 0.266745 # Ni6+2 O_3 H_
3 fourier 122.883342 0.300235 0.267331 0.266745 # H_ O_3 H_
Pair Coeffs
1 0.015000 2.524807 # Ni6+2 Ni6+2
2 0.044000 2.571134 # H_ H_
3 0.060000 3.118146 # O_3 O_3
Atoms
1 1 1 0.00000 10.66000 -10.90380 -10.06580
2 1 2 0.00000 11.99020 -10.38620 -7.90340
3 1 3 0.00000 11.04120 -10.55960 -8.06080
4 1 2 0.00000 10.57080 -9.80720 -7.65240
5 1 2 0.00000 11.45240 -8.37340 -10.14300
6 1 3 0.00000 10.73120 -8.88980 -10.55100
7 1 2 0.00000 10.82920 -8.75300 -11.51380
8 1 2 0.00000 13.10860 -10.72340 -11.06260
9 1 3 0.00000 12.70460 -11.15420 -10.28520
10 1 2 0.00000 12.94820 -12.09780 -10.35720
11 1 2 0.00000 10.76800 -11.99440 -12.47840
12 1 3 0.00000 10.27780 -11.25540 -12.07040
13 1 2 0.00000 9.33380 -11.45520 -12.22680
14 1 2 0.00000 10.50100 -13.04340 -8.61060
15 1 3 0.00000 10.58580 -12.91640 -9.57620
16 1 2 0.00000 9.85260 -13.42840 -9.96780
17 1 2 0.00000 8.37680 -9.70620 -9.79500
18 1 3 0.00000 8.61620 -10.65260 -9.84020
19 1 2 0.00000 8.21940 -11.05620 -9.04480
Bonds
1 1 1 3
2 1 1 6
3 1 1 9
4 1 1 12
5 1 1 15
6 1 1 18
7 2 2 3
8 2 3 4
9 2 5 6
10 2 6 7
11 2 8 9
12 2 9 10
13 2 11 12
14 2 12 13
15 2 14 15
16 2 15 16
17 2 17 18
18 2 18 19
Angles
1 1 3 1 15
2 1 6 1 9
3 1 18 1 3
4 1 18 1 12
5 1 9 1 15
6 1 18 1 15
7 1 18 1 9
8 1 12 1 15
9 1 18 1 6
10 1 3 1 6
11 1 6 1 12
12 1 3 1 12
13 1 3 1 9
14 1 6 1 15
15 1 9 1 12
16 2 1 3 2
17 3 2 3 4
18 2 1 3 4
19 2 1 6 5
20 3 5 6 7
21 2 1 6 7
22 2 8 9 1
23 2 1 9 10
24 3 8 9 10
25 2 1 12 13
26 2 1 12 11
27 3 11 12 13
28 2 16 15 1
29 3 16 15 14
30 2 1 15 14
31 2 1 18 19
32 2 1 18 17
33 3 19 18 17
data.K+H2O6 (4.46 KB)
data.Ni2+H2O6 (4.46 KB)
H2O.txt (603 Bytes)
in.K+H2O6 (1.13 KB)
in.Ni2+H2O6 (1.14 KB)
log.K+H2O6 (6.9 KB)
log.Ni2+H2O6 (13.7 KB)
param.qeq1 (200 Bytes)
param.qeq2 (202 Bytes)