Hello LAMMPS users,
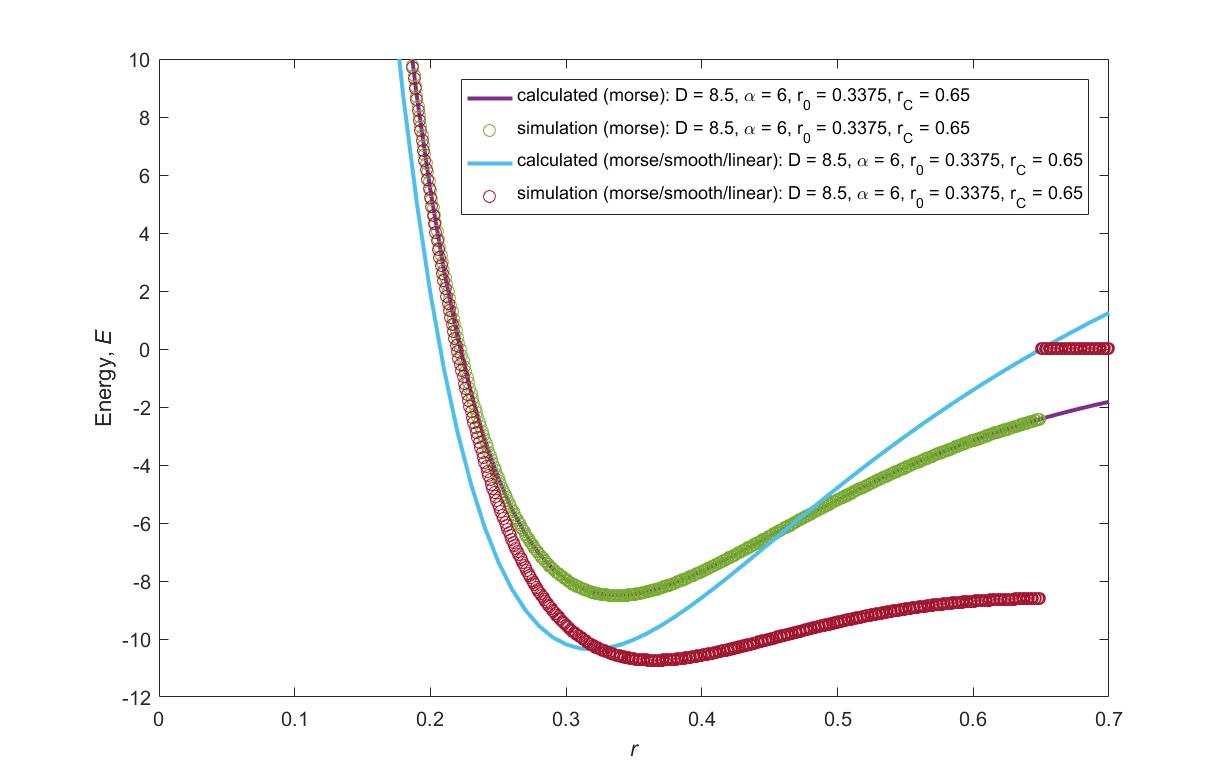
I have some problems with the morse/soft/linear potential. Doing a 2-bead simulation it seems I cannot reproduce the energy curve (E®) outlined by the equation here: https://lammps.sandia.gov/doc/pair_morse.html. Just as a sanity check I tried to reproduce the energy curve of the standard morse potential in lammps and the lammps output matched the energy equation for this case. You can see everything I have mentioned in the attached plot. If you notice in the lammps output for the morse/smooth/linear potential, the potential does not go to zero at the cutoff as it should. I also attached an animation of the simulation for clarity. I tested this with the latest stable version of LAMMPS, 22Aug18. I have attached input script as well. The simulation was ran on a single processor.
Thanks,
John

in.mem (58.5 KB)
From the plot it seems to me that the offset is not being applied to the morse/soft/linear potential, because the energy curve near the cutoff is horizontal, so it seems like the force goes to zero, but the energy doesn’t.
I might have made an error in the implementation, I will take a look later today. It might be a simple sign error but I am not sure.
Assuming you mean morse/smooth/linear, it seems like the cut-off is not properly applied even though it is set. Strangely enough, the output from pair_write looks normal and the code in single looks identical to the one in compute. I will have a closer look later.
So interestingly enough, if you change the neighbor skin distance, the symptoms of the problem also change. The randomness of it all leads me to suspect that some stuff is being used while not intialized or something. pair lj/smooth/linear does not have the same issues so I think it is related to lj/smooth/linear.