Dear lammps-users and developers
I would like to study the tensile process of A/B interface. The initial model consists of 24 A layers and 24 B layers.
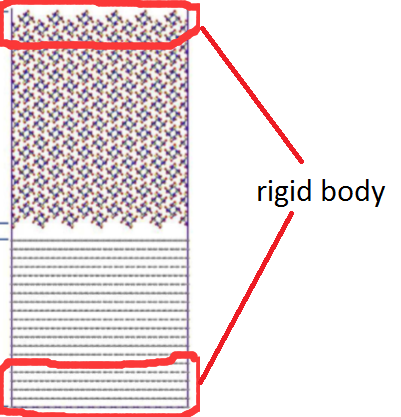
For the tensile test, I set the last six B layers (group base0) and three A layers (group additive0) as rigid body. And then 24 A layers are moved up 0.3 Å by 0.3 Å, used by command displace_atoms.
The problem I met is, it can work in LAMMPS 2009 version. However LAMMPS 2012 version and the later versions, it cannot work. The position of A layers(group additive0) is not moved. This perplexes me with a long time.
I need your help.
Best wishes
Hang Fan
Here is my script
in.create
clear
units real
dimension 3
atom_style full
read_data ${File}
include BX_field.in
group base0 molecule 1
group base1 molecule 2
group base union base0 base1
group additive1 molecule 3
group additive0 molecule 4
group additive union additive0 additive1
group bound union base0 additive0
group body union base1 additive1
variable t equal temp
variable e equal etotal
variable ep equal pe
variable ek equal ke
variable z0 equal xcm(base0,z)
variable z1 equal xcm(additive0,z)
timestep 0.5
thermo 1
thermo_style custom step temp etotal pe ke v_z0 v_z1
minimize 0 0 10000 1000000
velocity body create 600 428751 dist gaussian
fix bound bound rigid group 2 base0 additive 0 force * off off off torque * off off off
fix md body nvt temp 300 300 100 drag 0.2
(fix md body nvt 300 300 100 drag 0.2, Lammps 2009 Version)
run 10000
dump xyz all xyz 100 ${File}.0.xyz
run 0
undump xyz
in.main
variable File index BX.data
variable n loop 30
include in.create
label start_loop
fix print all print 1 "$t e {ep} {ek} {z0} {z1}" file {File}.$n.result
run 10000
unfix print
dump xyz all xyz 100 ${File}.$n.xyz
run 0
undump xyz
displace_atoms additive move 0 0 0.3 units box
next n
jump main.in start_loop
variable n delete