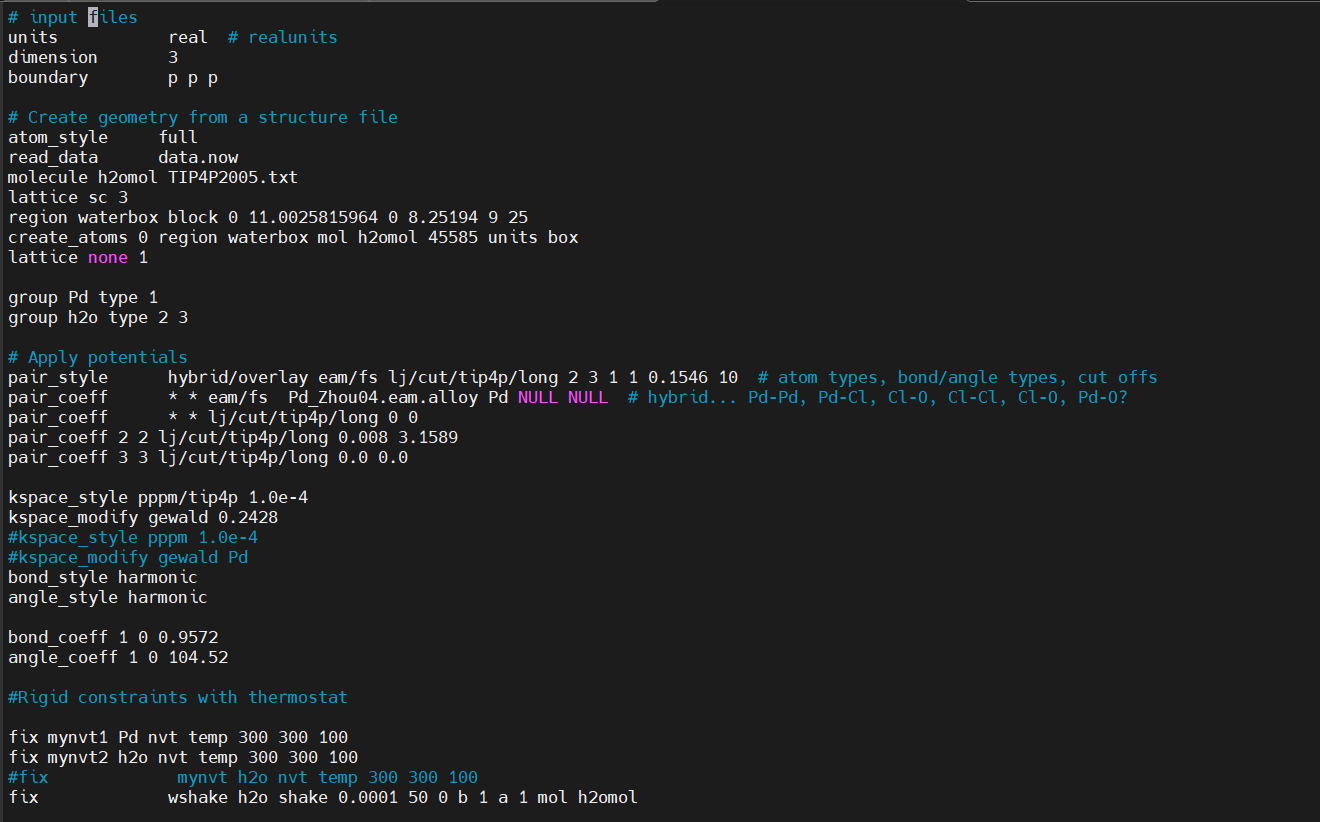
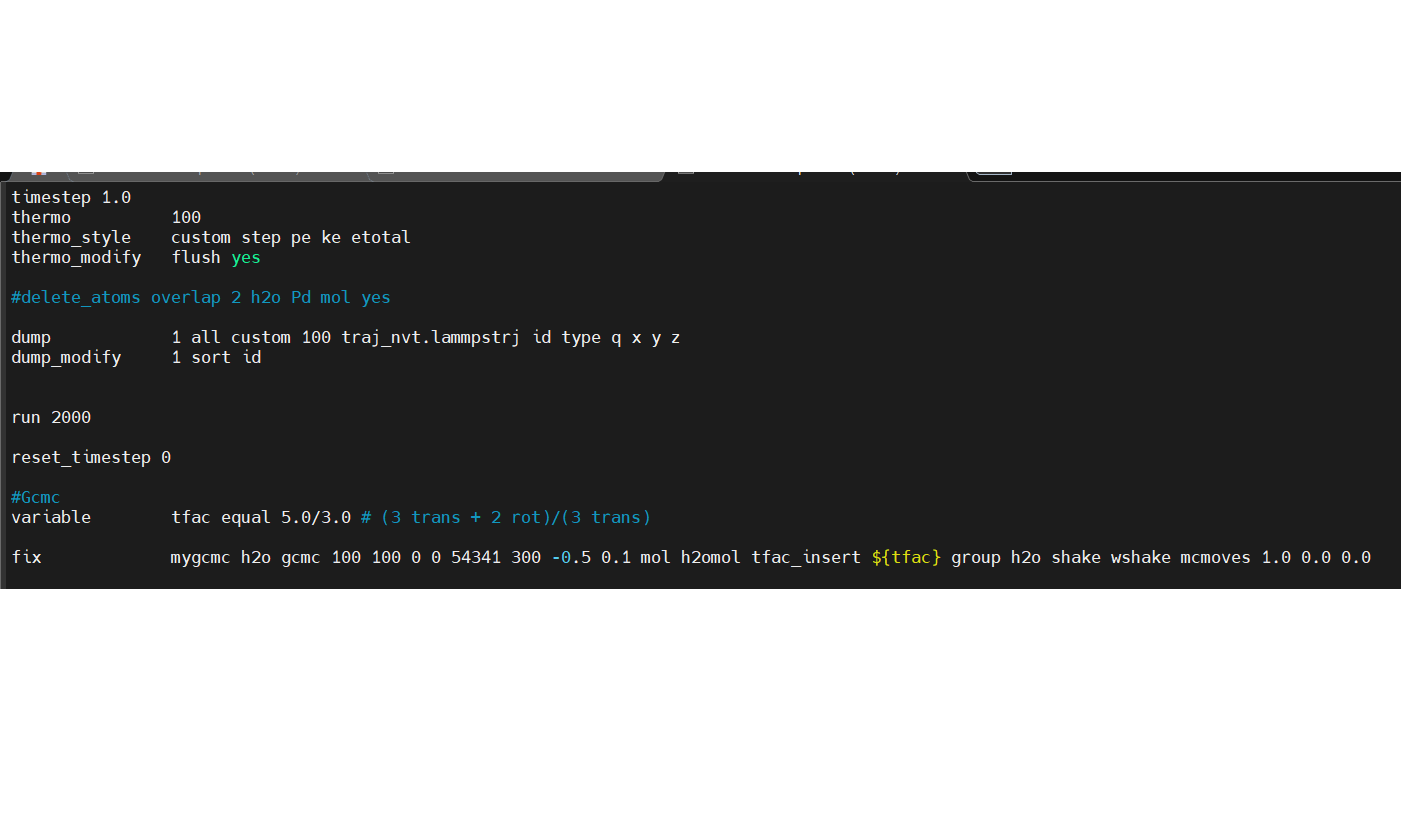
I recently just started learning lammps to run gcmc.
I managed to write the input script, but I have ran into issues like : ERROR: fix gcmc does currently not support full_energy option with molecules on more than 1 MPI process. (…/fix_gcmc.cpp:728)
Not really. What fix gcmc does is rather complex and convoluted. The fact that LAMMPS uses domain decomposition for parallelization is making matters more complicated for MC moves.
The complication comes from applying MC moves to molecules. If you can do without (i.e. only want insertions/deletions), then the latest version of LAMMPS will allow you to run in parallel with MPI, otherwise only multi-thread parallelization is available.