Hello lammps users,
I am new to lammps. I have two questions.
I have a system with around 40000 atoms. Will it take significantly less time if I run my simulation using mpi_run instead of serial_run?
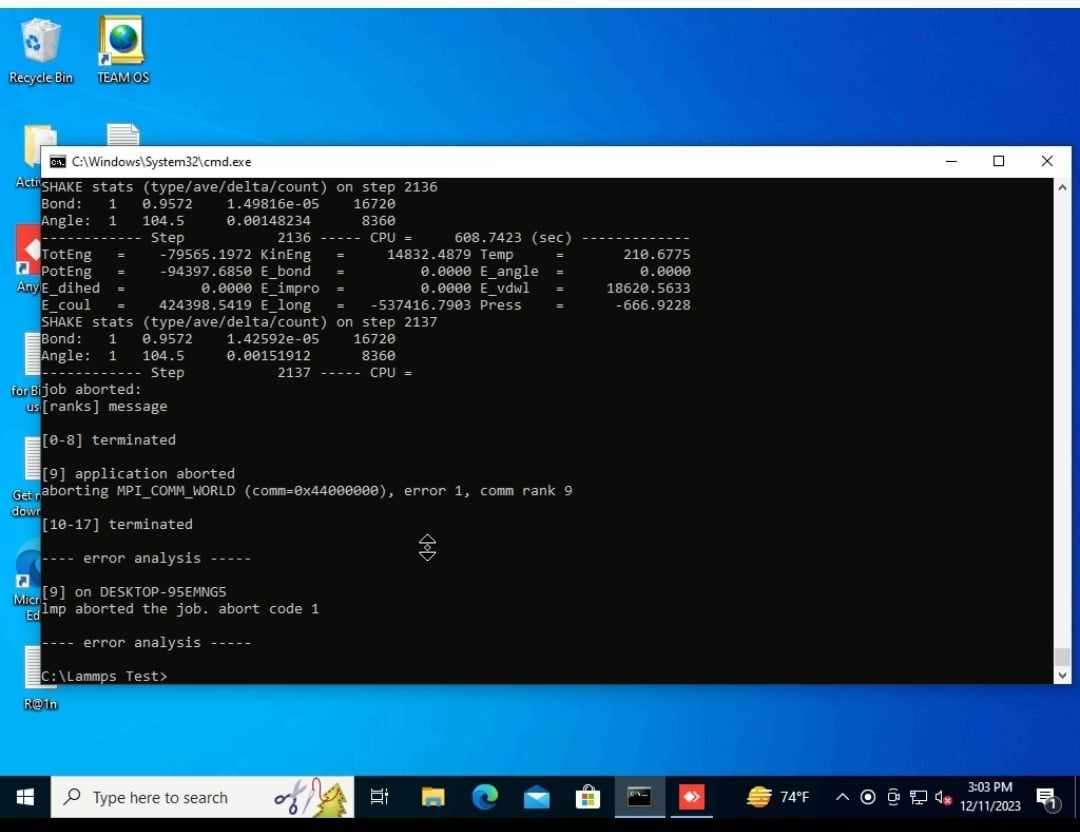
As I am running using the command “mpiexec -np 18 lmp -in NaCl.in” , I am constantly getting following error as described in the image during energy minimization step.
I am trying to simulate crumpled graphene electrode with NaCl electrolyte using constant charge method. I am running lammps ms-mpi executable 2 August 2023 version. I am attaching my data file and input file for reference. NaCl.txt (3.6 KB) soln100.lmpdat (2.1 MB)
Yes. Exact speedup depends on the details of your system and how many MPI processes you are using. Generally, you want to use only real hardware cores, not hyperthreading cores.
Some speedup, but not quite as efficient, can also be had from OpenMP multi-threading through setting OMP_NUM_THREADS and appending -sf omp to the command line. In some cases, a combination of threads and MPI is the fastest.
To know for certain, you should check out the “performance” “timings” blocks of the output https://docs.lammps.org/Run_output.html
for different choices of threads and MPI processes.
It is difficult to say what is causing the issue. Particularly since when I am running the attached files, I get a different output.
I am using the development version on Linux, but there is nothing that has been changed in the relevant parts of the code that can justify the difference, and the OS should not matter either. We do extensive tests on a variety of platforms (some you may not even have heard of) to verify that LAMMPS produces the same output (within the limits of floating point math accuracy) everywhere.
That said, I can easily run on windows, if need be to confirm my findings, but I am very confident (and we are talking about 15 years of experience with LAMMPS here) that this is not making a difference.
I am constantly getting that abort error around 2000-3000 steps using the same input script and structure as mentioned above. I am attaching a log file below of the run I am getting this error.
I tried with a smaller system of 1 M NaCl electrolyte with the same electrode but lesser electrolyte thickness which ran fine, but in this one I am constantly getting this error. Is that error due to any discrepancy in my system or code?
I have made electrolyte using avogadro and packmol, converted pdb to lammps data file using openbabel. I have made graphene sheet using Gopy package and crumpled it using eric hahn’s code attached below.
Then I assembled and attached charges to my electrode and electrolyte manually using excel.
I am running the code in a 18 core windows server using the command “mpiexec -np 18 lmp -in NaCl.in”.
I may sound desperate because I searched a lot and it seems to me that no one gets into this problem.
Thanks a lot again.
LAMMPS rarely/never aborts without an error message, but sometimes it doesn’t get flushed out to the screen. You need to figure out what that error message is, if LAMMPS is crashing. Does the input run correctly on 1 MPI rank?
You can try to get a “core” file and then read it with the gdb debugger tool. You may next to set ulimit -c unlimited if you are on Linux.
I ran your input again with some small adjustments:
there is no need to use “newton off” it slows things down
there is no need to output stuff every step as it slows things down, too.
thermo_modify lost ignore and thermo_modify lost/bond ignore hide errors and must not be used for a system like yours. With them you would probably have seen a ERROR: Lost atoms: original 31980 current 31976 or similar.
The system you simulate has serious problems. Your temperature rises to extreme levels (like over 10000K) which is an indication of a very bad starting structure or a very bad force field or both.
You cannot just ignore this. Running a molecular system at such high temperature will lead to all kinds of irreversible changes and render the entire simulation bogus.
I am convinced the reason you don’t get an error message is due to your suppression of the lost atoms or bonds errors. Suppressing a symptom does not solve a problem. At some point LAMMPS notices bogus data and just crashes because the checks that your data has become bogus are disabled.
The best process to figure out how to set up and equilibrate such a system properly is to set up small calculations of the individual subsystems and figure out how to setup such systems properly and run a stable simulation for that subsystem. That is much faster and you reduce concurrent sources of errors. I think this starts by trying to figure out where the high potential energy comes from and how it can be reduced before the first minimization. If that doesn’t work, you will have to do a sequence where you alternate multiple times between several 1000 steps of minimization and short MD runs that stop before temperature gets beyond 300K. Then quench again and repeat until the system can run without huge amounts of kinetic energy being generated. You may also need to reduce the timestep during that period.
You also need to discuss the scientific validity of your overall setup (force field and geometry), what I see looks questionable to me, but I am not an expert in this domain of research and those issues are not LAMMPS issues, but scientific issues and thus off-topic for this category.