Hi Lammps users,
Had taken a single organic molecule. Had converted the pdb file into .LAMPDAT file using OPEN BABEL.
Had used the following code.
LAMMPS data file generated by OpenBabel
26 atoms
26 bonds
42 angles
58 dihedrals
0 impropers
4 atom types
6 bond types
10 angle types
15 dihedral types
0 improper types
-5.40900 4.43900 xlo xhi
-5.40900 4.43900 ylo yhi
-5.40900 4.43900 zlo zhi
Masses
1 12.0107 # C
2 1.00794 # H
3 14.0067 # N
4 15.9994 # O
Atoms
1 1 1 0.00000 2.43400 -0.74400 -0.07300 # C
2 1 1 0.00000 2.13800 0.58400 0.25800 # C
3 1 4 -0.82000 3.10900 1.41100 0.74700 # O
4 1 2 0.41000 3.93900 0.91700 0.77900 # H
5 1 1 0.00000 0.84700 1.06700 0.08100 # C
6 1 2 0.41000 0.63900 2.10100 0.32200 # H
7 1 1 0.00000 -0.15800 0.23800 -0.42600 # C
8 1 1 0.00000 -1.57400 0.75400 -0.57100 # C
9 1 1 0.00000 -2.43000 0.50200 0.69500 # C
10 1 2 0.41000 -1.85600 0.83200 1.56500 # H
11 1 3 0.00000 -2.79700 -0.89800 0.85700 # N
12 1 2 0.41000 -2.82300 -1.14900 1.83600 # H
13 1 1 0.00000 -4.03500 -1.29000 0.19300 # C
14 1 2 0.41000 -4.90900 -0.68500 0.49100 # H
15 1 2 0.41000 -4.25000 -2.33800 0.41300 # H
16 1 2 0.41000 -3.92600 -1.20200 -0.89200 # H
17 1 2 0.41000 -3.32500 1.15000 0.63400 # H
18 1 4 -0.82000 -1.51200 2.15900 -0.84000 # O
19 1 2 0.41000 -2.40800 2.51000 -0.84000 # H
20 1 2 0.41000 -2.04600 0.23700 -1.41700 # H
21 1 1 0.00000 0.14900 -1.08100 -0.75800 # C
22 1 2 0.41000 -0.62100 -1.73600 -1.14800 # H
23 1 4 -0.82000 3.74300 -1.12800 0.13700 # O
24 1 2 0.41000 3.88000 -2.03000 -0.16700 # H
25 1 1 0.00000 1.44200 -1.57300 -0.57900 # C
26 1 2 0.41000 1.67800 -2.60100 -0.83900 # H
Bonds
1 2 10 11 # C: H
2 2 3 8 # C: H
3 2 23 26 # C: H
4 6 16 17 # O: H
5 5 16 10 # O: C
6 2 2 7 # C: H
7 1 3 2 # C: C
8 1 3 4 # C: C
9 1 2 1 # C: C
10 1 10 4 # C: C
11 1 10 18 # C: C
12 1 4 5 # C: C
13 6 14 15 # O: H
14 5 14 1 # O: C
15 1 1 6 # C: C
16 1 5 6 # C: C
17 2 5 9 # C: H
18 2 23 24 # C: H
19 2 23 25 # C: H
20 3 21 23 # N: C
21 5 12 6 # O: C
22 2 18 19 # C: H
23 3 21 18 # N: C
24 2 18 20 # C: H
25 6 12 13 # O: H
26 4 21 22 # N: H
Angles
1 9 14 1 2 # O: C: C
2 1 6 1 2 # C: C: C
3 9 14 1 6 # O: C: C
4 2 3 2 7 # C: C: H
5 2 1 2 7 # C: C: H
6 1 3 2 1 # C: C: C
7 2 2 3 8 # C: C: H
8 2 4 3 8 # C: C: H
9 1 4 3 2 # C: C: C
10 1 10 4 3 # C: C: C
11 1 5 4 3 # C: C: C
12 1 10 4 5 # C: C: C
13 1 6 5 4 # C: C: C
14 2 4 5 9 # C: C: H
15 2 6 5 9 # C: C: H
16 1 5 6 1 # C: C: C
17 9 12 6 1 # O: C: C
18 9 12 6 5 # O: C: C
19 10 16 10 11 # O: C: H
20 2 4 10 11 # C: C: H
21 2 18 10 11 # C: C: H
22 9 16 10 4 # O: C: C
23 9 16 10 18 # O: C: C
24 1 18 10 4 # C: C: C
25 5 6 12 13 # C: O: H
26 5 1 14 15 # C: O: H
27 5 10 16 17 # C: O: H
28 2 10 18 19 # C: C: H
29 7 21 18 10 # N: C: C
30 2 10 18 20 # C: C: H
31 8 21 18 19 # N: C: H
32 6 20 18 19 # H: C: H
33 8 21 18 20 # N: C: H
34 3 23 21 18 # C: N: C
35 4 23 21 22 # C: N: H
36 4 18 21 22 # C: N: H
37 6 26 23 24 # H: C: H
38 6 26 23 25 # H: C: H
39 8 21 23 26 # N: C: H
40 6 25 23 24 # H: C: H
41 8 21 23 24 # N: C: H
42 8 21 23 25 # N: C: H
Dihedrals
1 9 11 16 10 17 # H: O: C: H
2 6 4 16 10 17 # C: O: C: H
3 6 18 16 10 17 # C: O: C: H
4 7 7 3 2 8 # H: C: C: H
5 2 1 3 2 8 # C: C: C: H
6 2 4 3 2 7 # C: C: C: H
7 1 1 3 2 4 # C: C: C: C
8 2 10 3 4 8 # C: C: C: H
9 2 5 3 4 8 # C: C: C: H
10 1 10 3 4 2 # C: C: C: C
11 1 5 3 4 2 # C: C: C: C
12 13 14 2 1 7 # O: C: C: H
13 2 6 2 1 7 # C: C: C: H
14 12 14 2 1 3 # O: C: C: C
15 1 6 2 1 3 # C: C: C: C
16 2 3 10 4 11 # C: C: C: H
17 2 5 10 4 11 # C: C: C: H
18 12 16 10 4 3 # O: C: C: C
19 12 16 10 4 5 # O: C: C: C
20 1 3 10 4 18 # C: C: C: C
21 1 5 10 4 18 # C: C: C: C
22 7 19 10 18 11 # H: C: C: H
23 11 21 10 18 11 # N: C: C: H
24 7 20 10 18 11 # H: C: C: H
25 13 16 10 18 19 # O: C: C: H
26 14 16 10 18 21 # O: C: C: N
27 13 16 10 18 20 # O: C: C: H
28 2 4 10 18 19 # C: C: C: H
29 10 21 10 18 4 # N: C: C: C
30 2 4 10 18 20 # C: C: C: H
31 1 6 4 5 3 # C: C: C: C
32 2 3 4 5 9 # C: C: C: H
33 1 6 4 5 10 # C: C: C: C
34 2 10 4 5 9 # C: C: C: H
35 5 2 1 14 15 # C: C: O: H
36 5 6 1 14 15 # C: C: O: H
37 1 5 1 6 2 # C: C: C: C
38 12 12 1 6 2 # O: C: C: C
39 12 14 1 6 5 # O: C: C: C
40 15 12 1 6 14 # O: C: C: O
41 1 1 5 6 4 # C: C: C: C
42 12 12 5 6 4 # O: C: C: C
43 2 1 5 6 9 # C: C: C: H
44 13 12 5 6 9 # O: C: C: H
45 4 18 23 21 26 # C: C: N: H
46 8 22 23 21 26 # H: C: N: H
47 4 18 23 21 24 # C: C: N: H
48 8 22 23 21 24 # H: C: N: H
49 4 18 23 21 25 # C: C: N: H
50 8 22 23 21 25 # H: C: N: H
51 5 1 6 12 13 # C: C: O: H
52 5 5 6 12 13 # C: C: O: H
53 3 23 18 21 10 # C: C: N: C
54 4 10 18 21 22 # C: C: N: H
55 4 23 18 21 19 # C: C: N: H
56 8 22 18 21 19 # H: C: N: H
57 4 23 18 21 20 # C: C: N: H
58 8 22 18 21 20 # H: C: N: H
dimension 3
units metal
atom_style full
bond_style harmonic
dihedral_style none
boundary p p p
read_data INPUT.LMPDAT
pair_style lj/cut 1.25
pair_coeff * * 0.00650462 4
bond_coeff * 100.0 0.110
neighbor 0.4 bin
neigh_modify every 2 delay 10 check yes page 1000000
timestep 5.0
compute msd all msd average yes
fix msd all ave/time 1 1 1 c_msd[4] file msddumpfile
run 200
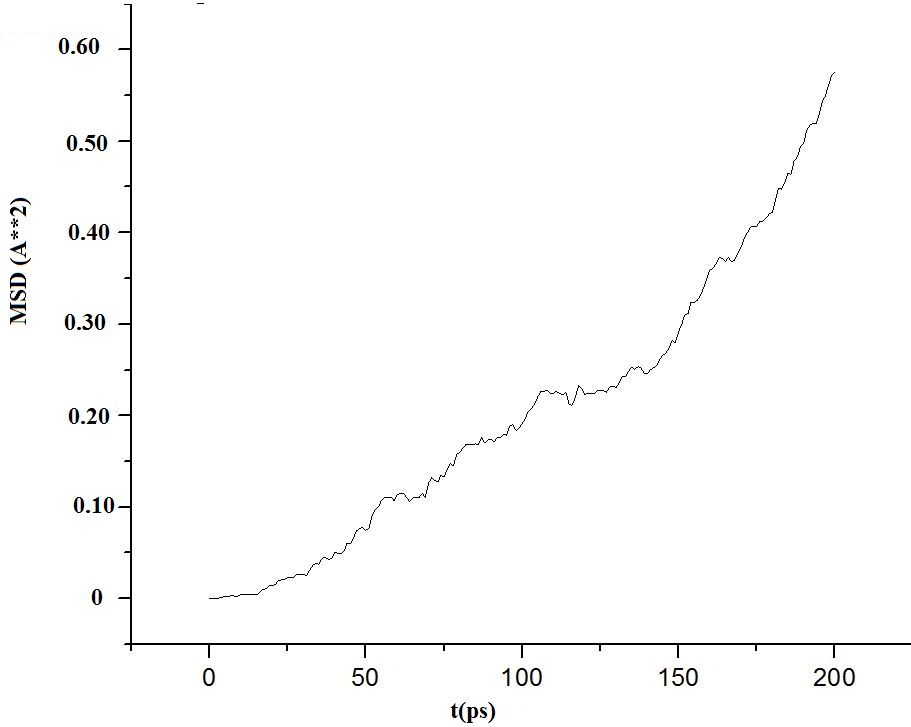
Have attached the graph (MSD Vs time). Is it correct?
Literature survey says that a straight line must be obtained. But the graph is not smooth. How to fix it?
Thanks,
Subashini.K