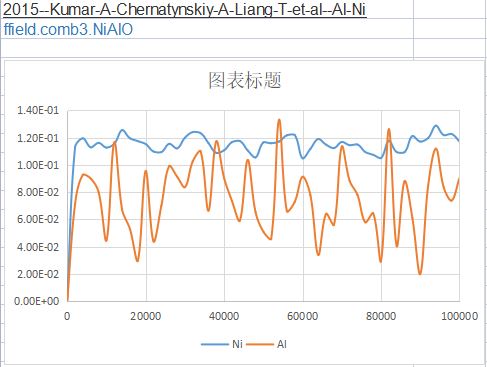
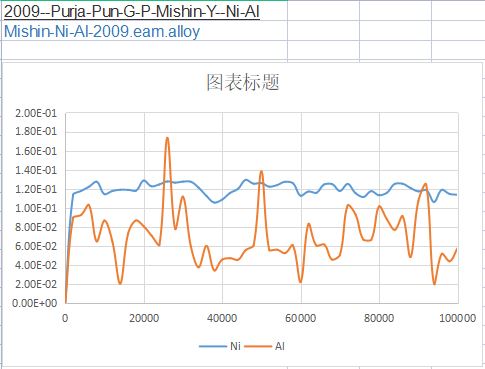
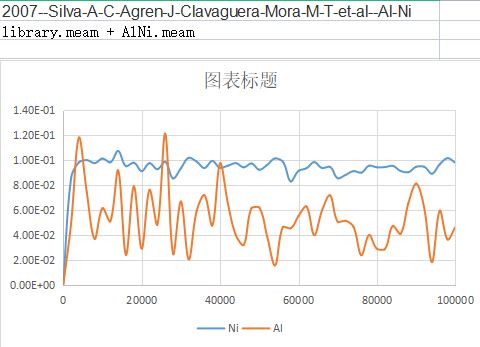
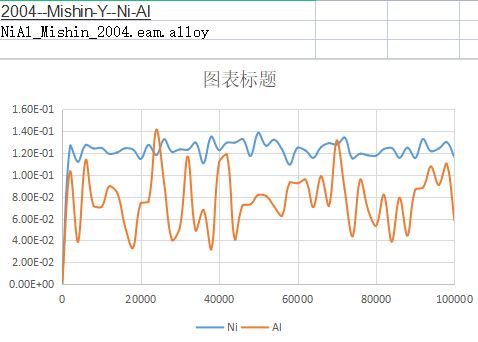
In order to calculate the diffusions, I want to calculated the MSD curve of Ni and Al in FCC Ni-1at%Al。
The code was set as follows:
#================== Start=================
#-----------------
units metal
boundary p p p
timestep 0.001
#----------------
#----- Al content
variable x_Al equal 0.01
#----- Diffusion Temperature
variable T_init equal 1200
#----------------
lattice fcc 3.58
region box block 0 5 0 5 0 5
create_box 2 box
create_atoms 1 box
#-----------------replace Ni with Al------------
set type 1 type/ratio 2 ${x_Al} 666
mass 1 58.6934
mass 2 26.981538
group Ni type 1
group Al type 2
#-----------------
pair_style meam/c
pair_coeff * * library.meam Al Ni AlNi.meam Ni Al
#-----------------
min_style cg
minimize 1e-15 1e-15 5000 5000
#-----------------
velocity all create ${T_init} 876848 dist gaussian
#------------------
fix 1 all npt temp ${T_init} ${T_init} $(100*dt) iso 0.0 0.0 1.0 drag 0.2
thermo 2000
run 20000
unfix 1
reset_timestep 0
#------------------ MSD calculation ---------
fix 1 all nvt temp ${T_init} ${T_init} $(100*dt)
compute 1 Ni msd com yes
variable twopoint_Ni equal c_1[4]/(6 * step * dt+1e-6)
fix 8 all vector 10 c_1[4]
variable fitslope_Ni equal slope(f_8)/(6 * 10 * dt)
compute 2 Al msd com yes
variable twopoint_Al equal c_2[4]/(6 * step * dt+1e-6)
fix 9 all vector 10 c_2[4]
variable fitslope_Al equal slope(f_9)/(6 * 10 * dt)
thermo_style custom step temp press vol c_1[4] c_2[4] v_twopoint_Ni v_fitslope_Ni v_twopoint_Al v_fitslope_Al
dump 2 all custom 1000 MSD_${T_init}.xyz id type x y z
variable t equal temp
variable st equal step
variable MSD_Single_Ni equal c_1[4]
variable MSD_Single_Al equal c_2[4]
fix 3 all print 100 “${st} ${t} ${MSD_Single_Ni} ${twopoint_Ni} ${fitslope_Ni}” file MSD_${T_init}_Ni.result screen no
fix 4 all print 100 “${st} ${t} ${MSD_Single_Al} ${twopoint_Al} ${fitslope_Al}” file MSD_${T_init}_Al.result screen no
run 100000
#================== END =================
The potential was obtained from DOI:10.1016/j.calphad.2006.02.006
However, the calculated MSD values kept around a certain values, not increased with increasing step.
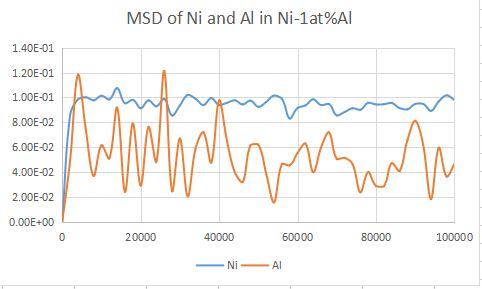
What should I do to correct the code and get a single increasing curve?
Thanks.
Zhenbang Wei
2021-07-20