Hello everyone,
I have a simulation with 1000 time steps(to try). If I use the MSD command as follows:
compute MSD all msd
fix result all print {intDump} "(step) $(c_MSD[4])" file “./datos/dump/msd.dump” screen no
I get the following output:
Fix print output for fix result
1 5.8497307081053128683e-24
2 2.3379300022448075844e-23
3 5.2503752979565054526e-23
4 9.3066253786508429407e-23
5 1.4484886146200516336e-22
6 2.0759348329274675512e-22
.
etc…
1000
I can graph this , however , it has just noise.
To improve this and because the fluid is in equilibrium , it occurs to me:
I would like to generate such files to every 100 steps of time. MSD restarting the counter in multiples of 100. Example , in step = 100 restart MSD to make t0 = 100 . And so on up to 1000 steps .
Thus would generate 10 files msd.dump : msd100.dump , msd200.dump , etc …
I do this to generate 10 straight lines of MSD vs. time , where time is from 0 to 100 in each graph. With these 10 graphs I want to get an average graph and calculate the diffusion coefficient .
Check the documentation : http://lammps.sandia.gov/doc/Section_howto.html#howto_22 however , I see, it is to calculate the value but not for the MSD vs. time graph with a good statistic.
as I can reset the MSD command every 100 steps to carry out what I have in mind? No where to start. I’m trying to do something with the if command but has not given me results .
Any help is welcome.
Regards
Using multiple time windows to generate better MSD stats is
common in MD. Unfortunately it is hard to do on-the-fly,
in the sense that it would require the code to store lots
of starting points (sets of coordinates for all atoms).
That’s easy for a user to abuse and blow-out memory.
So we haven’t implemented it. But you can easily
do it post-processing, e.g. from a dump file.
Steve
I have one doubt,
It does not intend to make the statistics on the fly. I wish every 100 time steps will create a file using msd.dump " uncompute " every 100 steps , so would be replacing the initial time positions uncompute invoke the command , so there would be no memory overhead .
Something like:
if ( step is multiple of 100 ) then
-> print to a file 100 msd calculations obtained with the current compute MSD.
-> uncompute the current MSD ( frees memory and forget the previous initial positions )
-> instantiate a new compute MSD ( New initial positions )
Using current functionality lammps Can I do something like this ?
Similar to what you would do object-oriented programming.
Christopher
You should be able to do what you outline using a loop in
your input script - see the jump and next commands.
To get the MSD values to a file, you will need to use
fix ave/time or a print statement.
My earlier email assumed you wanted to compute MSD
over long timescales and have multiple starting points
during the long time. That is a more typical way to
get good MSD stats. MSD for just 100 timesteps is
not likely to be an interesting value for most systems.
Steve
1 Like
thanks will try what you mentioned with loop and jump .
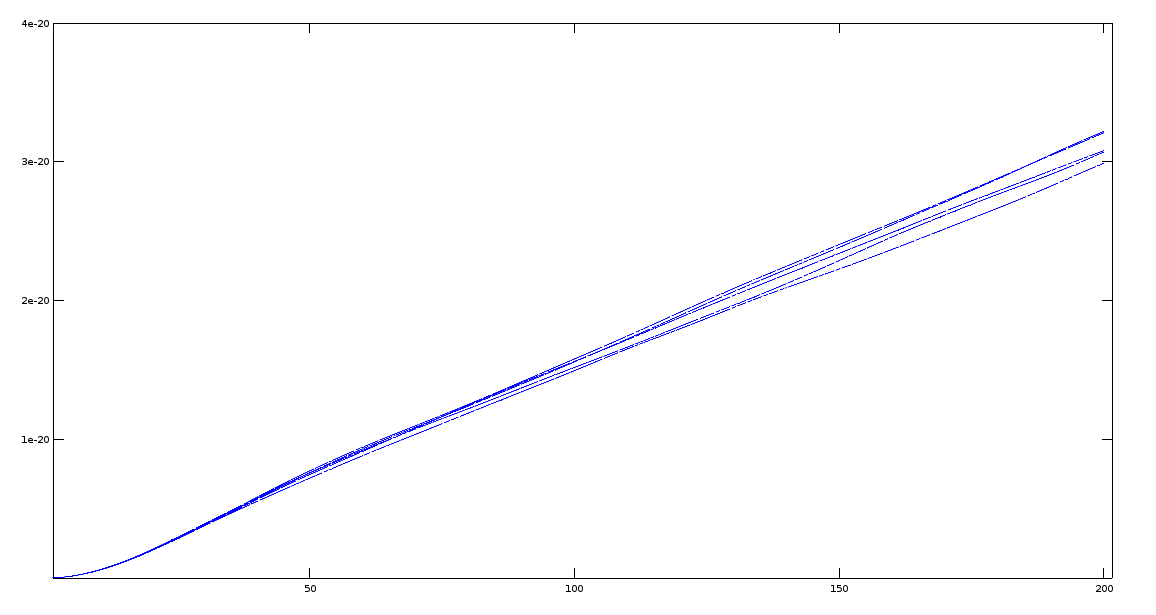
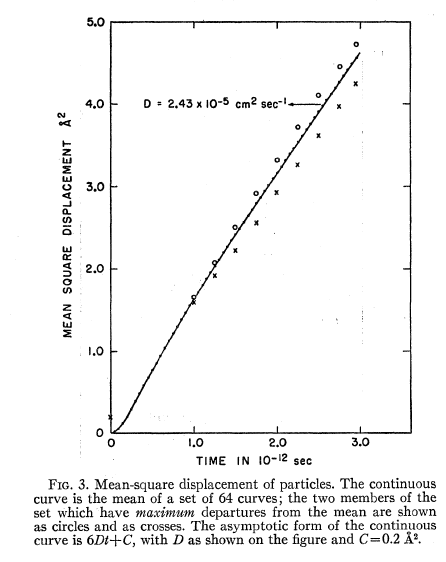
Actually what I want to draw 60 or more curves and averaged MSD with a post -processing. Similar to what was done in 1964 rahman .
I want to extract these curves one phase space trajectory , creating them at different times of origin. 100 steps is just one example , could generate 60 curves each of 1000 steps .
attached cover rahman 1964 .
The loop should be able to write out 60+ files each with
MSD values for an N-step run within the loop.
Note that image flags are important when computing an MSD.
LAMMPS uses them internally to unwrap the atoms when computing
an MS.
That
info needs to be in a dump file if you want to process it
for MSD thru rerun.
Steve
1 Like
thanks, loop worked for me . The MSD calculations were performed using rerun .
for example, the following code genre me 5 curves:
variable a loop 5
label loop
compute MSD all msd
fix result all print 1 “(step) (c_MSD[4])” file ./datos/dump/msd_rerun.dump.$a screen no
thermo 1
thermo_style custom step temp etotal ke pe epair
variable ini equal (($a-1)*200+1)
variable fin equal ($a*200)
rerun config.dump first {ini} last {fin} dump x y z ix iy iz vx vy vz
uncompute MSD
unfix result
next a
jump SELF loop
Now I can be parameterized to generate 60 I want and then post- processing by averaging those curves into one. After 200 steps , for all curves MSD value is approx. 3 Angstrom^2 , which is what I expected.
attached a screenshot .
thanks for the help.