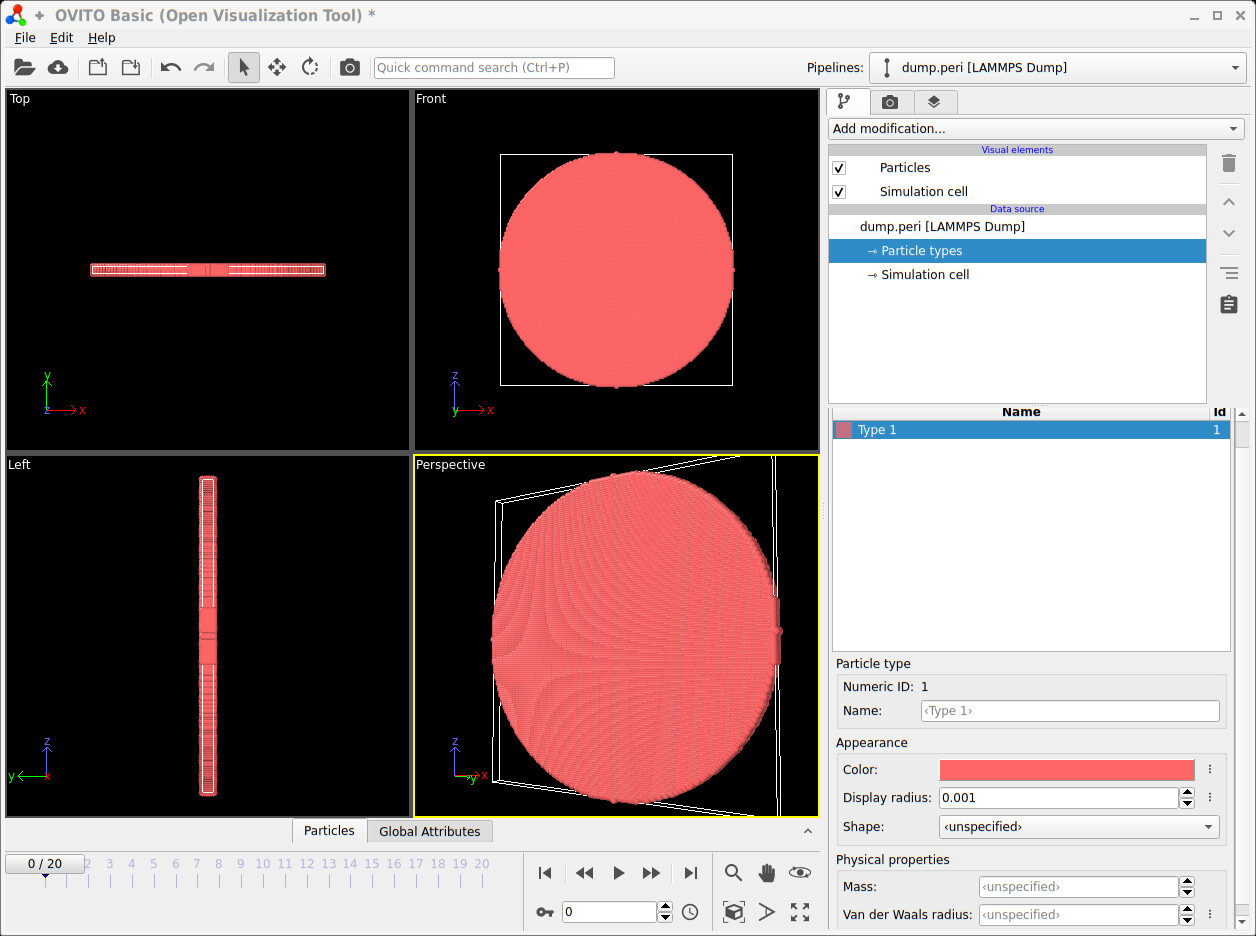
I’m attempting to simulate Peridynamics example in howto 8.5.9(10.5.9. Peridynamics with LAMMPS — LAMMPS documentation), but I think my result is something wrong.
<Linux abuntu 22.04, lammps 2Aug, 2023 version>
Only a simple spherical object is seen, such as howto, rather than breaking the plate.
when I put the dump.peri file in ovito, Videos like the howto example did not play. Am I missing something, or am I setting the system wrong?
my input file
3D Peridynamic simulation with projectile"
units si
dimension 3
boundary s s s
atom_style peri
atom_modify map array
neighbor 0.0010 bin
lattice sc 0.0005
Create desired target
region target cylinder y 0.0 0.0 0.037 -0.0025 0.0 units box
Make 1 atom type
create_box 1 target
Create the atoms in the simulation region
create_atoms 1 region target
pair_style peri/pmb
pair_coeff * * 1.6863e22 0.0015001 0.0005 0.25
Set mass density
set group all density 2200
volume = lattice constant^3
set group all volume 1.25e-10
Zero out velocities of particles
velocity all set 0.0 0.0 0.0 sum no units box
Use velocity-Verlet time integrator
fix F1 all nve
Construct spherical indenter to shatter target
variable y0 equal 0.00510
variable vy equal -100
variable y equal “v_y0 + stepdtv_vy”
fix F2 all indent 1e17 sphere 0.0000 v_y 0.0000 0.0050 units box
Compute damage for each particle
compute C1 all damage/atom
timestep 1.0e-7
thermo 200
dump D1 all custom 100 dump.peri id type x y z c_C1
run 2000
my log.lammps!
LAMMPS (2 Aug 2023 - Update 3)
OMP_NUM_THREADS environment is not set. Defaulting to 1 thread. (src/comm.cpp:98)
using 1 OpenMP thread(s) per MPI task
3D Peridynamic simulation with projectile"
units si
dimension 3
boundary s s s
atom_style peri
atom_modify map array
neighbor 0.0010 bin
lattice sc 0.0005
Lattice spacing in x,y,z = 0.0005 0.0005 0.0005
Create desired target
region target cylinder y 0.0 0.0 0.037 -0.0025 0.0 units box
Make 1 atom type
create_box 1 target
Created orthogonal box = (-0.037 -0.0025 -0.037) to (0.037 0 0.037)
1 by 1 by 1 MPI processor grid
Create the atoms in the simulation region
create_atoms 1 region target
Created 103110 atoms
using lattice units in orthogonal box = (-0.0370074 -0.00250025 -0.0370074) to (0.0370074 2.5e-07 0.0370074)
create_atoms CPU = 0.009 seconds
pair_style peri/pmb
pair_coeff * * 1.6863e22 0.0015001 0.0005 0.25
Set mass density
set group all density 2200
Setting atom values …
103110 settings made for density
volume = lattice constant^3
set group all volume 1.25e-10
Setting atom values …
103110 settings made for volume
Zero out velocities of particles
velocity all set 0.0 0.0 0.0 sum no units box
Use velocity-Verlet time integrator
fix F1 all nve
Construct spherical indenter to shatter target
variable y0 equal 0.00510
variable vy equal -100
variable y equal “v_y0 + stepdtv_vy”
fix F2 all indent 1e17 sphere 0.0000 v_y 0.0000 0.0050 units box
Compute damage for each particle
compute C1 all damage/atom
timestep 1.0e-7
thermo 200
dump D1 all custom 100 dump.peri id type x y z c_C1
run 2000
CITE-CITE-CITE-CITE-CITE-CITE-CITE-CITE-CITE-CITE-CITE-CITE-CITE
Your simulation uses code contributions which should be cited:
- PERI package for Peridynamics: doi:10.1016/j.cpc.2008.06.011
@Article{Parks08,
author = {M. L. Parks and R. B. Lehoucq and S. J. Plimpton and S. A. Silling},
title = {Implementing Peridynamics Within a Molecular Dynamics Code},
journal = {Comput.\ Phys.\ Commun.},
year = 2008,
volume = 179,
number = 11,
pages = {777–783}
}
CITE-CITE-CITE-CITE-CITE-CITE-CITE-CITE-CITE-CITE-CITE-CITE-CITE
Generated 0 of 0 mixed pair_coeff terms from geometric mixing rule
Neighbor list info …
update: every = 1 steps, delay = 0 steps, check = yes
max neighbors/atom: 2000, page size: 100000
master list distance cutoff = 0.0025001
ghost atom cutoff = 0.0025001
binsize = 0.00125005, bins = 60 3 60
2 neighbor lists, perpetual/occasional/extra = 1 1 0
(1) pair peri/pmb, perpetual
attributes: half, newton on
pair build: half/bin/atomonly/newton
stencil: half/bin/3d
bin: standard
(2) fix PERI_NEIGH, occasional
attributes: full, newton on
pair build: full/bin/atomonly
stencil: full/bin/3d
bin: standard
Peridynamic bonds:
total # of bonds = 10009800
bonds/atom = 97.0788
Per MPI rank memory allocation (min/avg/max) = 399.4 | 399.4 | 399.4 Mbytes
Step Temp E_pair E_mol TotEng Press Volume
0 0 0 0 0 0 1.3698216e-05
200 7.1886936e+26 5.5938384e+08 0 2.0944303e+09 7.4707857e+13 1.3698216e-05
400 2.7910257e+28 2.4071103e+09 0 6.200562e+10 1.3350783e+15 2.9760308e-05
600 3.5233498e+28 1.6580207e+08 0 7.5402083e+10 8.6213257e+14 5.8178431e-05
800 3.5496386e+28 74476758 0 7.5872119e+10 5.2465403e+14 9.6314445e-05
1000 3.5515831e+28 34031824 0 7.5873197e+10 3.7464553e+14 0.00013495275
1200 3.5522023e+28 24469677 0 7.5876857e+10 2.7957244e+14 0.00018087712
1400 3.5522091e+28 15594698 0 7.5868126e+10 2.2645264e+14 0.00022330653
1600 3.5520919e+28 13952085 0 7.5863981e+10 1.9006007e+14 0.00026605634
1800 3.5520675e+28 11189264 0 7.5860697e+10 1.6344991e+14 0.00030936902
2000 3.5519273e+28 11784016 0 7.5858299e+10 1.4320565e+14 0.00035308904
Loop time of 241.488 on 1 procs for 2000 steps with 103110 atoms
100.0% CPU use with 1 MPI tasks x 1 OpenMP threads
MPI task timing breakdown:
Section | min time | avg time | max time |%varavg| %total
Pair | 227.95 | 227.95 | 227.95 | 0.0 | 94.40
Neigh | 9.7381 | 9.7381 | 9.7381 | 0.0 | 4.03
Comm | 0.056631 | 0.056631 | 0.056631 | 0.0 | 0.02
Output | 1.257 | 1.257 | 1.257 | 0.0 | 0.52
Modify | 1.8921 | 1.8921 | 1.8921 | 0.0 | 0.78
Other | | 0.5897 | | | 0.24
Nlocal: 103110 ave 103110 max 103110 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Nghost: 0 ave 0 max 0 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Neighs: 1.60289e+07 ave 1.60289e+07 max 1.60289e+07 min
Histogram: 1 0 0 0 0 0 0 0 0 0
FullNghs: 3.57082e+07 ave 3.57082e+07 max 3.57082e+07 min
Histogram: 1 0 0 0 0 0 0 0 0 0
Total # of neighbors = 35708150
Ave neighs/atom = 346.31122
Neighbor list builds = 95
Dangerous builds = 0
Total wall time: 0:04:02
thanks for your help!