Here is my code
----------------------------------------------
#-----initialization-----
units metal
dimension 3
boundary f f f
atom_style atomic
neighbor 2.0 bin
timestep 0.00001
#----geometric variable-----
variable tdamp equal 10
variable scale equal 1.0
variable file string fire_hydrant_with_90_twin
variable theta equal 20*3.1415926/180
#---------------------------
variable x_scale equal 140*${scale}
variable x_1 equal -90*${scale}
variable x_2 equal -61*${scale}
variable x_3 equal -45*${scale}
variable x_4 equal -14*${scale}
variable x_5 equal 12*${scale}
variable x_6 equal 33*${scale}
variable low_1 equal 105*${scale}
variable low_2 equal 87.9*${scale}
variable low_3 equal 168.54*${scale}
variable low_4 equal 250.02*${scale}
variable low_5 equal 332.34*${scale}
variable low_6 equal 415.5*${scale}
variable high_1 equal 45*${scale}
variable high_2 equal 45.9*${scale}
variable high_3 equal 42.54*${scale}
variable high_4 equal 40.02*${scale}
variable high_5 equal 38.34*${scale}
variable high_6 equal 37.5*${scale}
variable cone_hi equal 300*${scale}
#-----generation-----
lattice fcc 3.6150 orient x 1 1 1 orient y 1 1 -2 orient z -1 1 0
region sc prism -140 140 -140 140 -15 350 0.0 0.0 0.0 units box
create_box 3 sc
region model_1 prism -${x_scale} ${x_1} -140 140 0 350 0.0 0.0 0.0 units box
region model_2 prism ${x_1} ${x_2} -140 140 0 350 0.0 0.0 0.0 units box
region model_3 prism ${x_2} ${x_3} -140 140 0 350 0.0 0.0 0.0 units box
region model_4 prism ${x_3} ${x_4} -140 140 0 350 0.0 0.0 0.0 units box
region model_5 prism ${x_4} ${x_5} -140 140 0 350 0.0 0.0 0.0 units box
region model_6 prism ${x_5} ${x_6} -140 140 0 350 0.0 0.0 0.0 units box
region model_7 prism ${x_6} ${x_scale} -140 140 0 350 0.0 0.0 0.0 units box
create_atoms 1 region model_1
create_atoms 1 region model_3
create_atoms 1 region model_5
create_atoms 1 region model_7
#-----twin-----
lattice fcc 3.6150 orient x -1 -1 1 orient y 1 1 2 orient z -1 1 0
create_atoms 2 region model_2
create_atoms 2 region model_4
create_atoms 2 region model_6
#-----shape-----
region fuck_1 cone z 0 0 ${low_1} ${high_1} 0 ${cone_hi} side out units box
region fuck_2 cone z 0 0 ${low_2} ${high_2} 0 ${cone_hi} side out units box
region fuck_3 cone z 0 0 ${low_3} ${high_3} 0 ${cone_hi} side out units box
region fuck_4 cone z 0 0 ${low_4} ${high_4} 0 ${cone_hi} side out units box
region fuck_5 cone z 0 0 ${low_5} ${high_5} 0 ${cone_hi} side out units box
region fuck_6 cone z 0 0 ${low_6} ${high_6} 0 ${cone_hi} side out units box
delete_atoms region fuck_1
delete_atoms region fuck_2
delete_atoms region fuck_3
delete_atoms region fuck_4
delete_atoms region fuck_5
delete_atoms region fuck_6
lattice diamond 3.57
region top prism -210 0 -105 105 307 317 0.0 0.0 0.0 units box
create_atoms 3 region top
lattice fcc 3.6150 orient x 1 1 1 orient y 1 1 -2 orient z -1 1 0
region down prism -140 140 -140 140 -15 0 0.0 0.0 0.0 units box
create_atoms 1 region down
#-----mass-----
mass 1 63.55
mass 2 63.55
mass 3 12.0096 #carbon mass (amu)
#------ force field ------
pair_style hybrid eam/alloy tersoff lj/cut 3.615
pair_coeff * * eam/alloy Cu_zhou.eam.alloy Cu Cu NULL
pair_coeff * * tersoff SiC.tersoff NULL NULL C
pair_coeff 1*2 3 lj/cut 0.001034 2 3.615
#------ computation ------
compute peratom all pe/atom
#-----minimize-----
variable t equal "time"
variable temp equal "temp"
variable pe equal "pe"
group top1 type 3
group down1 region down
displace_atoms top1 rotate 0 0 0 0 1 0 20
fix dontgo all wall/reflect xlo -140 xhi 140 ylo -140 yhi 140 zlo -15 zhi 350
#dump 1 all custom 50 ${file}_mini_*.dump id type xu yu zu c_peratom
#dump_modify 1 sort id scale no
#thermo 50
#thermo_style custom step temp pe
#min_style cg
#minimize 1.0e-5 1.0e-5 1000 100000
#undump 1
#-----------------------
#------ stage 4 ------ DEFORMATION
fix freeze down1 setforce 0.0 0.0 0.0
fix top top1 setforce 0.0 0.0 0.0
fix 1 all nve
fix 2 all temp/berendsen 100.0 100.0 ${tdamp}
dump 8 all custom 500 ${file}_*.dump id type xu yu zu c_peratom
dump_modify 8 sort id
thermo 500
thermo_style custom step temp pe
run 100000
unfix 2
undump 8
#------ stage 4 ------ DEFORMATION
fix 2 all temp/berendsen 200.0 200.0 ${tdamp}
dump 8 all custom 500 ${file}_*.dump id type xu yu zu c_peratom
dump_modify 8 sort id
thermo 500
thermo_style custom step temp pe
run 100000
unfix 2
undump 8
#------ stage 4 ------ DEFORMATION
fix 2 all temp/berendsen 300.0 300.0 ${tdamp}
dump 8 all custom 500 ${file}_*.dump id type xu yu zu c_peratom
dump_modify 8 sort id
thermo 500
thermo_style custom step temp pe
run 100000
unfix 2
undump 8
#------ stage 4 ------ DEFORMATION
fix 2 all temp/berendsen 400.0 400.0 ${tdamp}
dump 8 all custom 500 ${file}_*.dump id type xu yu zu c_peratom
dump_modify 8 sort id
thermo 500
thermo_style custom step temp pe
run 100000
unfix 2
undump 8
#------ stage 4 ------ DEFORMATION
fix 2 all temp/berendsen 500.0 500.0 ${tdamp}
dump 8 all custom 500 ${file}_*.dump id type xu yu zu c_peratom
dump_modify 8 sort id
thermo 500
thermo_style custom step temp pe
run 100000
unfix 2
undump 8
write_restart ${file}_thermo.restart
#----------------------------------------------
dump 1 all custom 50 ${file}_mini_*.dump id type xu yu zu c_peratom
#dump_modify 1 sort id scale no
thermo 50
thermo_style custom step temp pe
min_style cg
minimize 1.0e-8 1.0e-8 1500 100000
undump 1
#------ stage 4 ------ DEFORMATION
fix 2 all temp/berendsen 500.0 500.0 ${tdamp}
restart 1000 restart_nt-Cu_sametemp_*.restart
dump 8 all custom 500 ${file}_*.dump id type xu yu zu c_peratom
thermo 500
thermo_style custom step temp pe etotal pxx pyy pzz fmax
run 100000
unfix 2
undump 8
write_restart ${file}_final.restart
However, there are -nan in the output
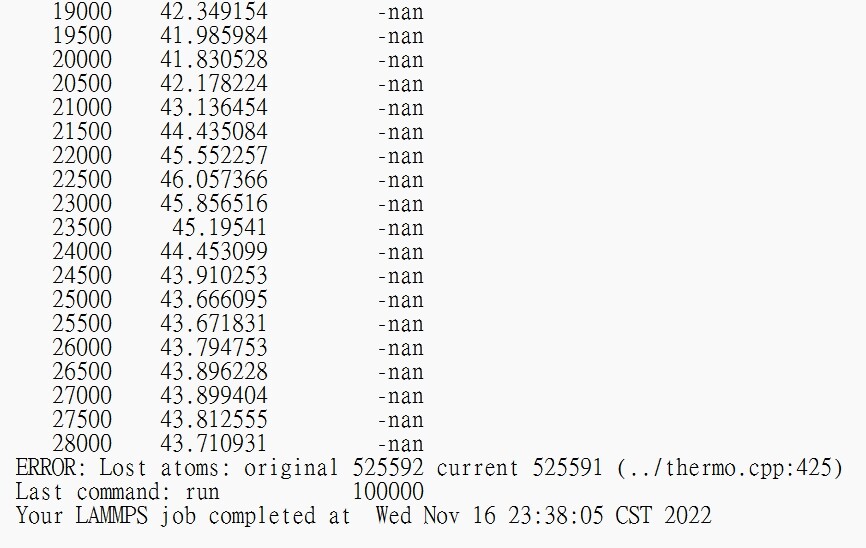
I know maybe there is a wrong potential
but I dont’t know how to check that