Thank you for developing and maintaining that code.
I am trying to calculate with PTFE (C2F4, bandgap ~ 5 eV) with this code.
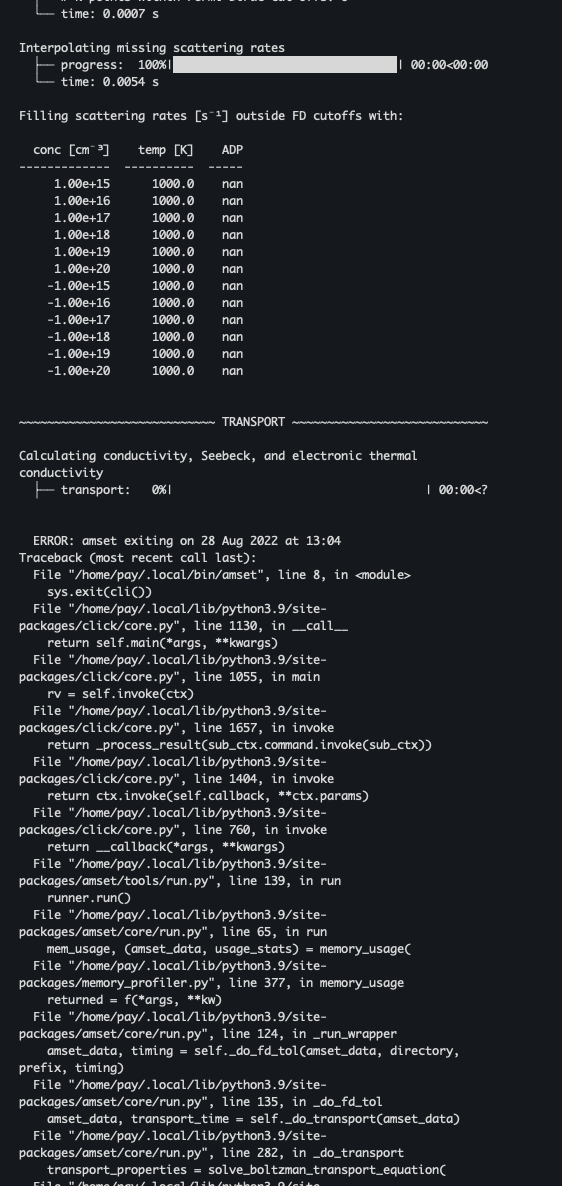
I keep getting NaN values while performing the ADP calculation. It seems that some singular value is generated during the calculation, but I can’t get a sense of it at all. How to solve it?
I have attached the error log, setting, and poscar screenshots.
If you explicitly upload the elastic script to the official website, many people seem to get help.
I confirmed that ADP is being calculated successfully with the script.
However, I noticed one strange thing. My structure has vdW interaction of 1D polymer.
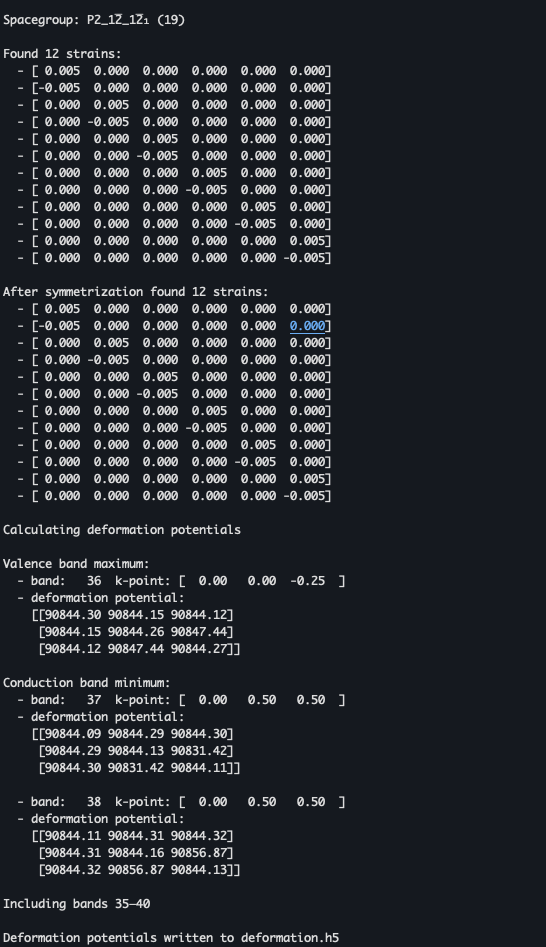
If we look at the deformation potential in other examples like silicon, we can see that it has a value of 1-10.
But, my result is 90000. No matter how bulk and vdW systems are different, the 90000 value seems too absurd (of course, the ADP mobility value is also too small).
There are two questions.
Isn’t the deformation value well defined in the vdW structure?
The functional dependency seems very large. Have you done any benchmarking on this?
Yeah that doesn’t like right at all. Are you sure all your calculations used consistent settings? The INCAR should be exactly the same for bulk and deformations.
Oh, yes ICORELEVEL is definitely needed in the undistorted structure as it is used as the reference level. That would definitely result in your massive deformation potentials.
The calculation has been performed very successfully. It is almost similar to the mobility of polymer materials reported in several literatures (~1 cm^2/Vs at 300K).
I have confirmed that AMSET works successfully on polymers as well.