Hello all
I tried to execute nanoindentation simulation for calculating hardness.
I reference several tutorials and I tried to set fixed bottom layer and heat bath layer on top of it.
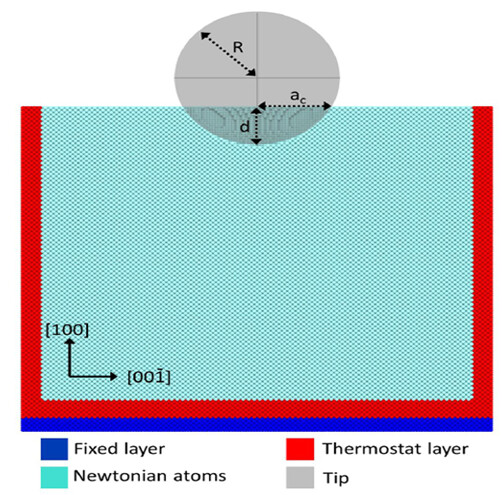
At first I used boundary condition p p s.
I used region command
region 3 block INF INF INF INF 142 145 units box
region 2 block INF INF INF INF INF 1.0 units box
region 7 block INF INF INF INF INF 3.0 units box
region 9 block INF INF INF INF 3 5 units box
group top region 3
group bottom region 2
group base region 7
group inner subtract all bottom top
group heatbath region 9
and to make fixed layer and heat bath layer I used command
####---------- totally fix all boundaries ---------------------
fix 7 base setforce 0.0 0.0 0.0
velocity base set 0 0 0
fix 9 heatbath langevin 300 300 1 699232
But the atoms located in bottom move during indentation simulation like this picture
(Bottom atoms are not fixed!)
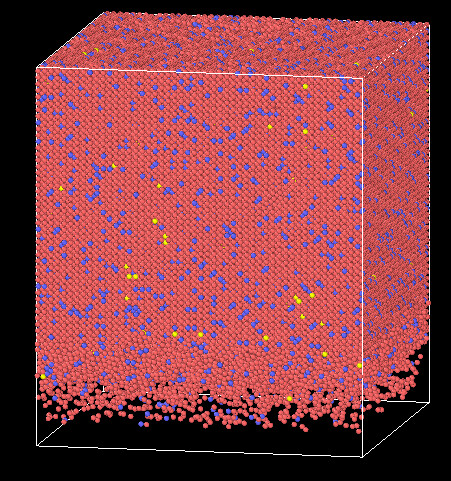
Could you give any advice for solving this problem please?
Thank you.
P.S. This is full script
# SGB under nanoindentation
# Iman Salehinia, Washington State University, Dec. 2014
#special thanks to Mark Tschopp for providing the GB structures
# ---------- Setup Variables ---------------------
variable etol equal 1.0e-25
variable ftol equal 1.0e-25
variable maxiter equal 5000
variable maxeval equal 10000
variable latparam equal 4.82
# ---------- Initialize Simulation ---------------------
clear
units metal
dimension 3
boundary p p s
atom_style atomic
# ---------- Create Atomistic Structure ---------------------
#lattice bcc ${latparam}
read_data "input.lmp" # Garrett
group Fe type 1
group Cr type 2
group W type 3
variable n_atoms equal atoms
variable zlow equal zlo
variable zhigh equal zhi
print "zlow = ${zlow}, zhigh = ${zhigh}"
#creating top and bottom partitions in order to trim the fat from the grains
region 3 block INF INF INF INF 142 145 units box
region 2 block INF INF INF INF INF 1.0 units box
region 7 block INF INF INF INF INF 3.0 units box
region 9 block INF INF INF INF 3 5 units box
group top region 3
group bottom region 2
group base region 7
group inner subtract all bottom top
group heatbath region 9
#creating the step in the material
#region stepatoms block INF INF 0.01 INF 106 INF units box
#group step region stepatoms
#delete_atoms group step
delete_atoms group top
delete_atoms group bottom
# ---------- Define Interatomic Potential ---------------------
pair_style hybrid/overlay eam/alloy eam/fs
pair_coeff * * eam/alloy FeCrW_d.eam.alloy Fe Cr W
pair_coeff * * eam/fs FeCrW_s.eam.fs Fe Cr W
neighbor 2.0 bin
neigh_modify delay 10 check yes
# ---------- Define Settings --------------------------------
compute csym all centro/atom bcc
compute eng all pe/atom
#this exports the data into the csp.all file
dump 101 all custom 1000 indent*.data id type x y z c_csym c_eng
#dump 101 all custom 1000 csp.all id type x y z c_csym c_eng
# ---------- Run Minimization ----------------------------
reset_timestep 0
thermo 10
#thermo_style custom step pe lx ly lz press pxx pyy pzz
min_style cg
minimize ${etol} ${ftol} ${maxiter} ${maxeval}
# ---------- totally fix all boundaries ---------------------
fix 7 base setforce 0.0 0.0 0.0
velocity base set 0 0 0
fix 8 heatbath langevin 300 300 1 699232
# ---------- initial velocities ---------------------------
compute new inner temp
velocity inner create 10.00 4882748 temp new
#fix 10 all nve
fix 4 all nvt temp 300.0 300.0 0.01 drag 1.0
#fix 4 all temp/rescale 5 0.01 0.01 0.005 0.05
# ---------- relaxation --------------------------------
timestep 0.001
thermo 100
thermo_modify temp new
thermo_style custom step temp
run 5000
# ---------- indentation -------------------------------------------
variable z equal "190-0.1*step*dt"
fix 5 all indent 100.0 sphere 72 72.0 v_z 30 units box
# ---------- Run with indenter ------------------------------------------
timestep 0.001
thermo 10
thermo_modify temp new
thermo_style custom step temp f_5[3]
run 300000