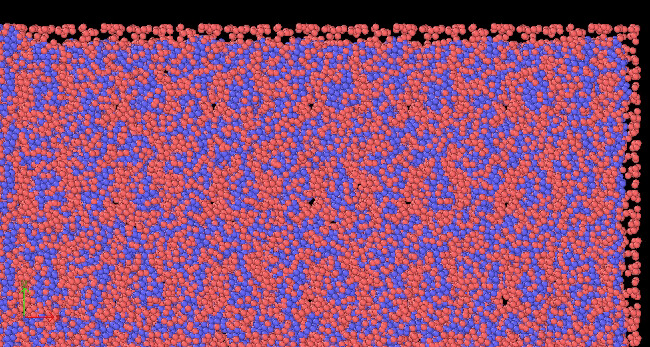
Hi, I am trying to run simulation of nanocutting of amorphous material CuZr by using diamond tool. The steps i have taken is to first prepare CuZr in a separate file, then generate datafile to be read into cutting input file.
Lammps version: LAMMPS 64-bit 27May2021
Procedure of preparing CuZr: minimization, melting, freezing, replication, relaxation.
Potentials I am using is MEAM for Cu-Zr, and Morse for Cu-C/Zr-C.
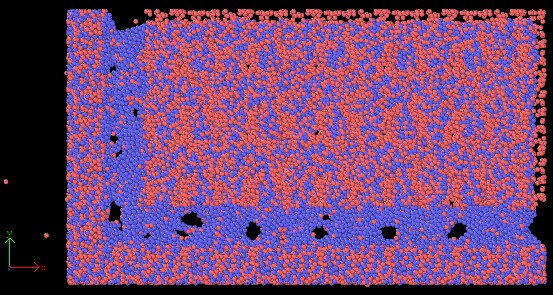
when I run my cutting input file, the workpiece(CuZr) atoms scatter right away in all directions.
I realised that there are some atoms overlapping between the boundaries.
There are three region: boundary, temp-layer and newton layer.
Workpiece has 100000 atoms, but the total number of atoms of the three regions is 100060.
I wonder if this extra 60 overlapped atoms caused this problem.
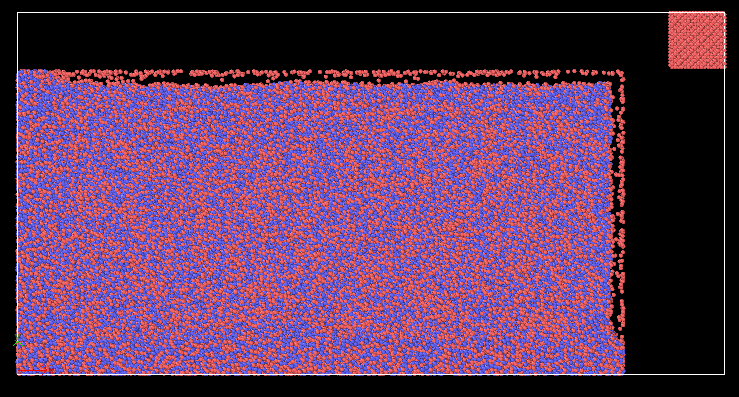
after minimization, workpiece looks weird as well.

after 10000 steps, lost atom
cutting.in (2.0 KB)
I have attached the cutting input file. The CuZr file is too big, so if it is needed, I could share a link later.
It would be great if anyone could provide some guidance. Thank you very much.