Hi,
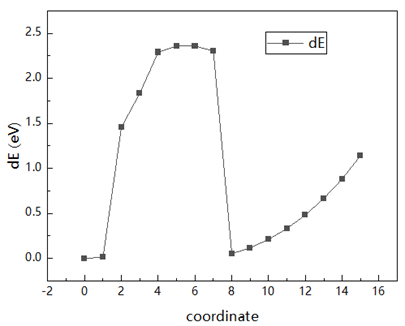
I am trying to calculating dislocation activation energy using LAMMPS NEB. I successfully obtained the energy change, but there are two energy maxima. I observed the changes in atomic positions using OVITO and speculated that the first maximum was due to dislocation nucleation, while the second maximum was due to dislocation slip.
I wanted to analyze only dislocation nucleation, so I do a second NEB calculation using replica 5 as the fianl file. ButThe final energy will still rise. May I ask what may be the cause of this?
Here is my input file:
#-------------------------------
units metal
boundary p s s
atom_style atomic
timestep 0.001#1fs
neighbor 2.0 bin
neigh_modify every 1 delay 0
atom_modify map array sort 0 0.0
variable u uloop 40
read_data initial.neb
pair_style eam
pair_coeff * * Ni_u3.eam
#----------------------------------------------
fix 1 all nve
fix 2 all neb 0.1 parallel ideal perp 3 end last/efirst 1
dump 1 all custom 1000 dump.neb.$u id type xu yu zu
min_style quickmin
neb 0 0.5 10000 10000 100 final final.neb
#-----------------------------------------------------------------------
Here are my first neb result and ovito results:
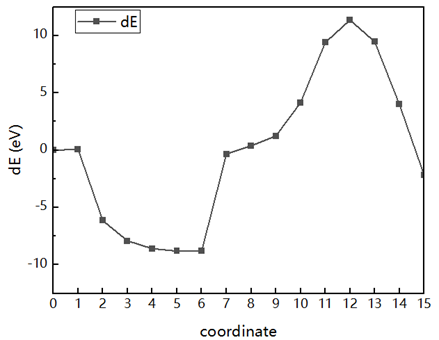
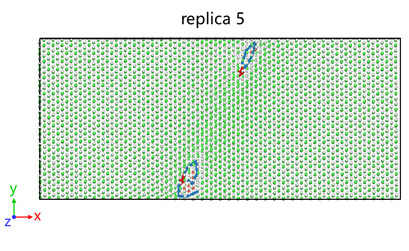
Here are the results of my second NEB: