zrO.dat (17.8 KB)
finial.data (32 Bytes)
in.zrO (1.3 KB)
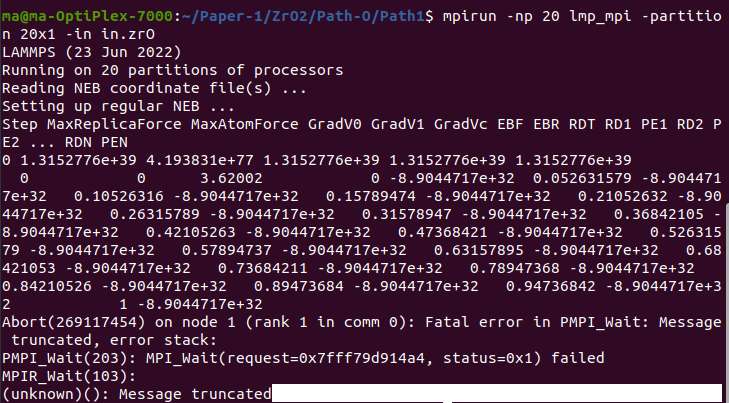
These are my caculated files, which resulted in a fatal error. But I can’t understand why it is like this, and I take some time still could not solve it. Could any one help me with it. Thank you so much! (Lammos version 23 Jun 2022)
Having values of 1e77 for what seems to be the MaxAtomForce is a good hint of why you may receive such an error.
Thank you for your help. Now I know maybe it’s due to the MaxAtomForce load in this error. I still don’t know why this force could be so big. 
Most likely your initial topology is wrong (some atoms may be too close?). You can try to perform minimization before starting NEB, it may solve the issue. If it does not solve the issue, have a look at your initial topology with VMD or Ovito, you may see that something is really wrong with it.
Thank you so much. Next, I will try to solve it with your suggestions.
Pengfei
Dear Simon:
Thanks for your suggestion. Now I can work it well done.
First, I change the initial topology that I increase the lattice length about 1.5 times. But it is seems made the MaxAtomforce more big than before.
Second, I applied the minimization for it. When I use “min_style fire” it still did’t work, so i used the “min_style quickmin”. It could be worked well. BUT! there still is a question. The each item number including petation energy looked like incorreced… 
Well I canno’t comment on the fact that the energy look incorrect, but what I can say is that the initial lattice parameter is usually chosen based on the expected equilibrium distance of the atoms. So the fact that you have to use an artificially increased value for the simulation to start without error is already a big warning.
Did you look at your system in VMD? Does it look good (i.e. no apparent overlap of the atoms, no big discontinuity near the periodic boundaries) ?
Thank you. Yes, I checked the system with Ovito. It seems like no problem.
I just made a supercell 4
45 for a CIF file from the ICSD website in the software Material Studio. Then I use this supercell for the above-mentioned NEB process.
I confused that do I still need to perform a equilibrium for the 4
44 supercell file so that reach to a resonable lattice parameter.
Pengfei
I have looked at your original data file. It looks suspicious since it has an odd number of atoms in it.
One would expect an even number. If I run the following input:
units metal
boundary p p p
dimension 3
atom_style charge
read_data zrO.dat
mass 1 91.224
mass 2 15.9994
pair_style buck/coul/long 10.0 10.0
pair_coeff * * 1e-30 1e-30 0.0
pair_coeff 1 2 1502.11 0.345 5.1
pair_coeff 2 2 9547.96 0.224 32.0
kspace_style pppm 0.0001
delete_atoms overlap 0.01 all all
write_data ZrO-no-overlap.data pair ij
I get a new data file with 478 atoms. Apparently there are two atoms in your data file that are closer than 0.01 Angstrom. This is all that is needed to produce unreasonable energies, forces, and pressures. However that does not seem to be sufficient. I need to increase the overlap cutoff to 0.5 angstrom and then 10 atoms get deleted and the energies and pressure look more reasonable.
But matters look worse when visualizing your initial geometry. It doesn’t look like a proper crystal when including a periodic image (left) and it becomes even more evident when looking at two atom type sublattices (right):
Obviously, two sides of the geometry have both, too many Zr and too many O atoms.
1 Like
Hi,Axel.
Thank you for your precious time. From your suggestions, I tried some examples. I found that this could work well with another original data file. Case 1: ICSD(CIF)–VESTA(xyz)–Ovito(lammps data file). Case 2: ICSD(CIF)–Ovito(lammps data file). Before, I used case 1 which could not run normally. So, I changed the method for the generation of the data file in Case 2, and it worked well.
Pengfei