Hi,
as I was preparing my EAM model driver for submission into the KIM repository I encountered a problem which I couldn't solve. It concerns small boxes and periodic boundary conditions. Maybe it's a trivial one, but currently I'm stuck.
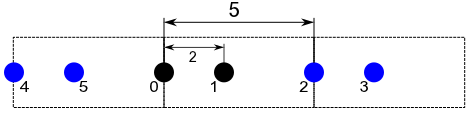
I attached a small 1D sketch which should describe the problem.
There is a simulation box with length 5 containing 2 atoms (index 0 and 1). They are sitting at x=0 and x=2, the cutoff is assumed to be 4.
The problem is the calculation of the electron density for the ghost atoms 2 and 5.
To calculate this for atom 2, one would need the contribution from atom 3. But this is not available since atom 2 is not contributing and has no neighbors. With periodic boundaries this however should not be a problem because atom 2 is the same as atom 0. So the contribution from 3 to 2 is the same as from 1 to 0. What happens with the KIM API and NEIGH_PURE and NEIGH_RVEC (half and full) neighbor lists is the following:
atoms 0 and 1 have the correct density assigned
atom 2 is missing the contribution from atom 3 (or 0)
atom 5 is missing the contribution from atom 4 (or 1)
This is not a problem for the energy, but the forces will come out wrong.
A simple solution is to map the neighbors back to the original atoms,
making the calculation of my example possible with only 2 atoms.
This means, that if I access atom 2 I actually am accessing atom 0.
Is there a way to "map" ghost atoms to real atoms? I am thinking of a function which gives you the contributing atom correspoding to a ghost atom. In my case something like an array which maps them back would be great so I could simply do
atom_rho[remap[2]] += some_value
where remap[2]=0, because atom 2 is the same as atom 0.
Or is there any other way to calculate the correct density for atoms 2 and 5 without knowing anything about the size of the simulation box and its geometry?
Any help is very much appreciated.
Best regards,
Daniel