Dear all,
I am reading the example file (lammps/examples/KAPPA/in.langevin) in the github.
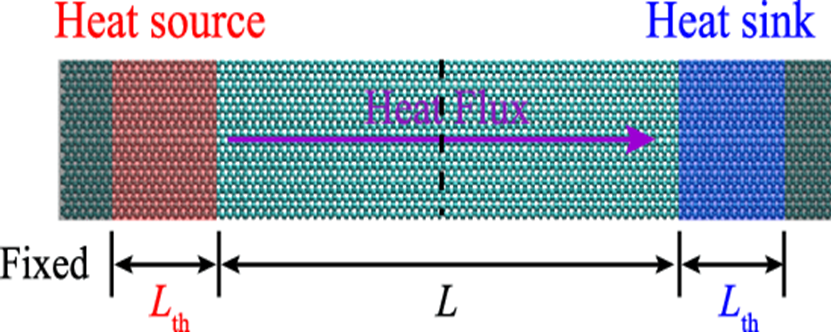
The image above shows a typical NEMD schematic.
Two Langevin thermostats, each at a different temperature, are applied to specific regions to create a temperature gradient.
From the example file:
region hot block INF INF INF INF 0 1
region cold block INF INF INF INF 10 11
fix 1 all nve
fix hot all langevin ${thi} ${thi} 1.0 59804 tally yes
fix cold all langevin ${tlo} ${tlo} 1.0 287859 tally yes
The regions for the hot and cold Langevin thermostats are defined. However, I noticed that the Langevin thermostat is applied to “all” atoms, regardless of whether they are in the hot or cold region.
Wouldn’t it make more sense to define groups for the hot and cold regions and apply the thermostats specifically to those groups, as shown below?
region hot block INF INF INF INF 0 1
region cold block INF INF INF INF 10 11
group g_hot block hot
group g_cold block cold
fix 1 all nve
fix hot g_hot langevin ${thi} ${thi} 1.0 59804 tally yes
fix cold g_cold langevin ${tlo} ${tlo} 1.0 287859 tally yes
Could anyone clarify if there’s a specific reason why the thermostats are applied to “all” atoms in the original script?