Dear LAMMPS experts,
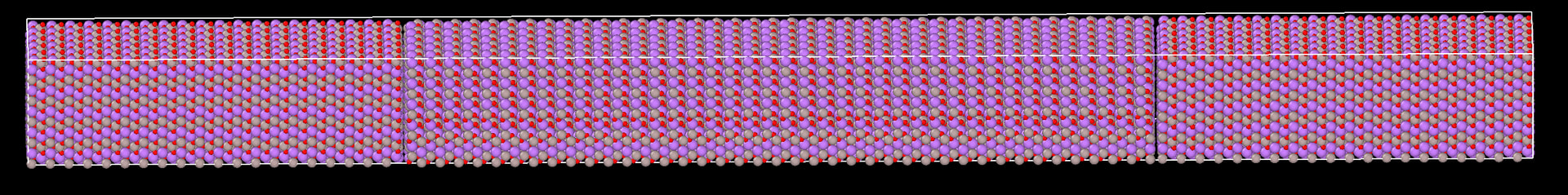
I am using heat add/subtract method to get a temperature profile across LiAlO2 using ReaxFF potentials for single crystal and bicrystal with grain boundary. Here, 0.0005 eV heat is added at the two ends/step and -0.001 ev/step heat is subtracted from the center. The shape is long in x direction as shown below:
LAMMPS data file written by OVITO Basic 3.5.4
38400 atoms
3 atom types
0.000000000000 418.410640000000 xlo xhi
0.000000000000 31.380798000000 ylo yhi
0.000000000000 31.633710000000 zlo zhi
Masses
1 6.94000000 # Li
2 26.98153860 # Al
3 15.99900000 # O
The code is below
#---------initialization---------
echo screen
units metal
dimension 3
boundary p p p
atom_style charge
read_data single_p1_x
variable t equal 300
variable p equal 200 # correlation length
variable s equal 10 # sample interval
variable d equal $p*$s
##############################
Force Field Parameters
##############################
pair_style reaxff lmp_control safezone 2.0 mincap 100
pair_coeff * * reaxff Li Al O
timestep 0.001
1st equilibration run
velocity all create 300 87287
region hotl block 0 10.41 INF INF INF INF
region hotr block 408 418.41 INF INF INF INF
region cold block 200 221 INF INF INF INF
compute Thot all temp/region hotl
compute Tcold all temp/region cold
fix 1 all nvt temp $t $t 0.001
fix 3 all qeq/reax 1 0.0 10.0 1e-6 param.qeq
thermo 100
run 10000
unfix 1
unfix 3
velocity all scale 300
2nd equilibration
reset_timestep 0
fix 1 all nve
fix 3 all qeq/reax 1 0.0 10.0 1e-6 param.qeq
fix hotl all heat 1 0.0005 region hotl
fix hotr all heat 1 0.0005 region hotr
fix cold all heat 1 -0.001 region cold
thermo_style custom step temp c_Thot c_Tcold
thermo 1000
run 10000
temperature profile
compute layers all chunk/atom bin/1d x lower 25 units box
fix 2 all ave/chunk 10 100 1000 layers temp file profile.heat
thermo 100
run 100000
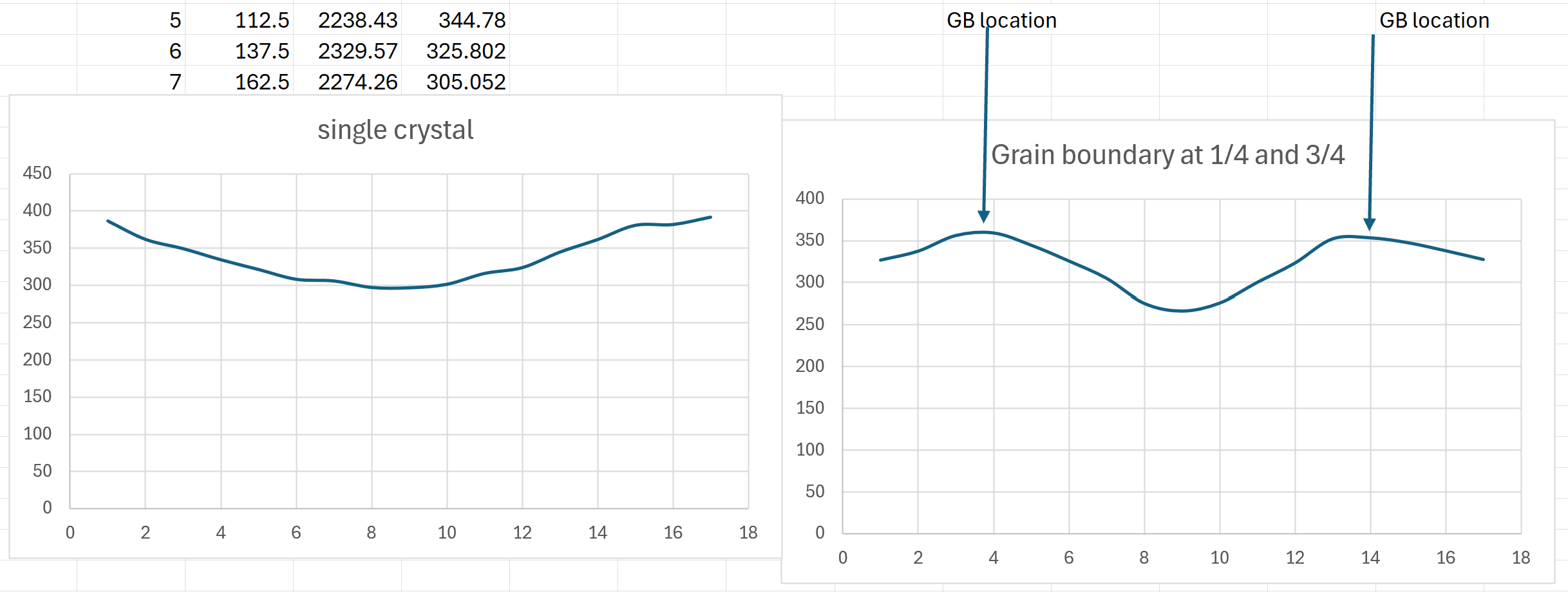
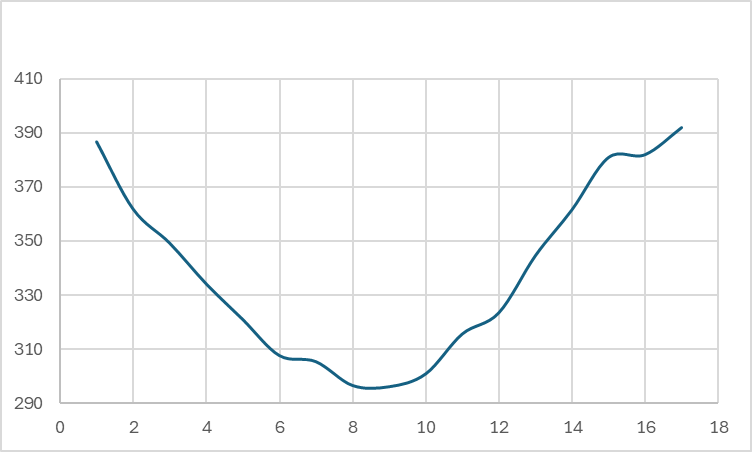
For the single crystal in x direction, I get a pretty linear profile shown below.
However, as soon as I put 2 grain boundaries at the 1/4 length and 3/4 length (to maintain symmetry), the same exact input file gives a very unexpected temperature profile with temperature spikes at the location of grain boundaries as shown below
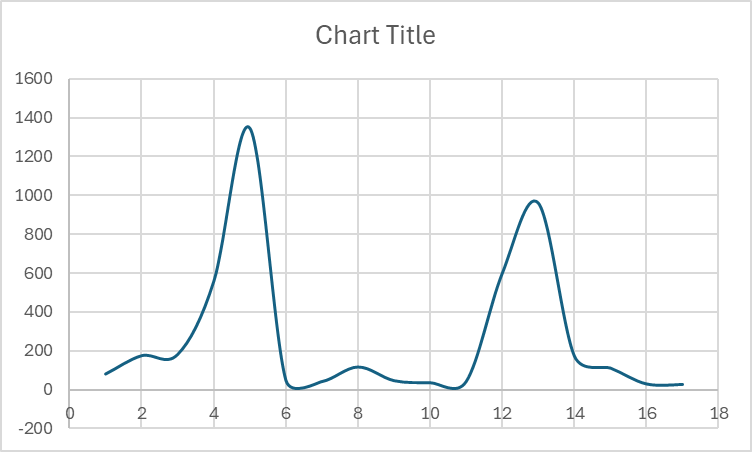
May I ask if I am doing something wrong here? I tried different grain boundary types like sigma 5 and sigma 41 and both are showing similar behavior. Is there any suggestions/corrections I can try please?
I highly appreciate any inputs from you.
Sincerely,
Ankit