Currently, I am practicing with subjecting a carbon nanotube to tension in order to induce fracture. I’m following an exercise from a book, but I have encountered a problem. Instead of the nanotube elongating, this is what’s happening:
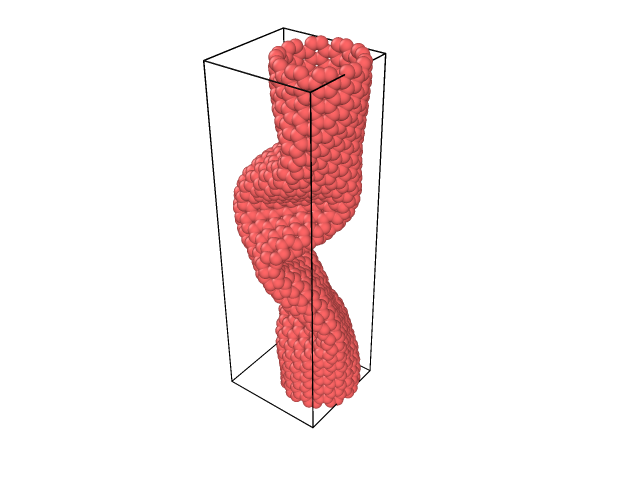
It seems to be compressing instead. I’ve tried altering the size of the simulation box, but it appears that this doesn’t change (In the script you will be able to see the size that I assigned to the box, which is larger than my nanotube, which does not appear when viewing it with OVITO), preventing the CNT from stretching. I’ve also tried fixing and unfixing the boundary limits, but I can’t find a solution to stretch it until it breaks.
Here’s the script I’m using:
#Script inspired by an exercise from the book "Computational Material Science by June Gunn Lee"
units metal
boundary f f p #PBC on z boundary only
#p: periodic, f:non-periodic & fixed, s:non-periodic & wrap
#wrap: face position always encompasses the atoms
atom_style atomic
#Structure
read_data r_d.data
mass 1 12.01
change_box all x final -100 100 y final -100 100 z final -150 150
#CENTER THE CNT
group g_cnt type 1
variable carbon_xcm equal -1*xcm(g_cnt,x)
variable carbon_ycm equal -1*xcm(g_cnt,y)
variable carbon_zcm equal -1*xcm(g_cnt,z)
displace_atoms g_cnt move ${carbon_xcm} ${carbon_ycm} ${carbon_zcm}
variable zmax equal bound(g_cnt,zmax)-4
variable zmin equal bound(g_cnt,zmin)+4
region top block INF INF INF INF ${zmax} 40 #78.593536
region bottom block INF INF INF INF -40 ${zmin}
group g_top region top
group g_bottom region bottom
group g_boundary union g_top g_bottom
group g_body subtract all g_boundary
#Potential
pair_style airebo 2.5 1 1
pair_coeff * * CH.airebo C
### Calculation of stress ####
compute body_temp g_body temp
compute_modify body_temp dynamic yes
compute body_pe all pe/atom
compute body_st all stress/atom body_temp pair bond
#compute body_st all stress/atom body_temp pair_bond
compute strAll all reduce sum c_body_st[3]
compute strC g_cnt reduce sum c_body_st[3]
#c_body_st[3]; compute body stress of zz in one column of array
variable szzC equal c_strC/(8.784*count(g_cnt))*(10^-4)
variable szz equal v_szzC
thermo 100
thermo_style custom step c_body_temp vol press lz v_szz
thermo_modify lost ignore norm yes
###NVE relaxation at 300K######
timestep 0.001
velocity g_body create 300 4928359 dist gaussian rot yes units box
velocity g_body zero linear
#zero; make g_body's momenta zero by velocity adjust
fix 1 all nve
fix 2 g_body temp/berendsen 300.0 300.0 0.1
fix 3 g_boundary setforce 0.0 0.0 NULL
#top and bottom's x, y fixed
dump relaxAll all custom 2000 relaxAll.dat id zs c_body_pe c_body_st[3]
fix 4 all ave/time 1 100 100 c_strAll file stress_relax_All.dat
#in stress_relax_All.dat file, z stress converges close to 0
# 1 atm = 1.01325 bar = 1.01325x10^5 Pa = 1.01325 x 10^5 N/m^2
dump snapshotrelax all xyz 5000 CNT_relax.xyz
run 10000
write_restart restart.10000
undump relaxAll
undump snapshotrelax
unfix 3
unfix 4
#Applying tension ###
reset_timestep 0
fix 3 g_boundary setforce 0.0 0.0 0.0
velocity g_bottom set 0.0 0.0 0.2 units box
velocity g_top set 0.0 0.0 -0.2 units box
#Tensional strain on both ends (top and bottom) of CNT
#strain = timestep*strain rate
#Lammps's atomic stress must be divided by atomic volume
# For crystal, atomic volume = atomic mass/density
#for solid deformation, need ionic radius or van der Waals readius
#initial system V (40.5 x 40.5 x 80 A^3)
dump tensileAll all custom 5000 CNT_tensile.dat id zs c_body_pe c_body_st[3]
dump snapshot all xyz 100 CNT_tensile.lammpstrj
fix 22 all ave/time 1 100 100 c_strAll file stress_tensile_Body.dat
#Total atomic stress of all atoms to have units of stress (pressure)
fix 33 all ave/time 1 100 100 v_szz file stress_tensile_Gpa.dat
run 60000
write_restart restart.60000