Dear developers and users,
Recently I started to use AMSET and trying to understand its functionality. I am trying to calculate the scattering rate and mobility for a 2D material. I generated wavefunction.h5 and deformation.h5 as per guidance from AMSET documentation. Below is the Run parameter setting.
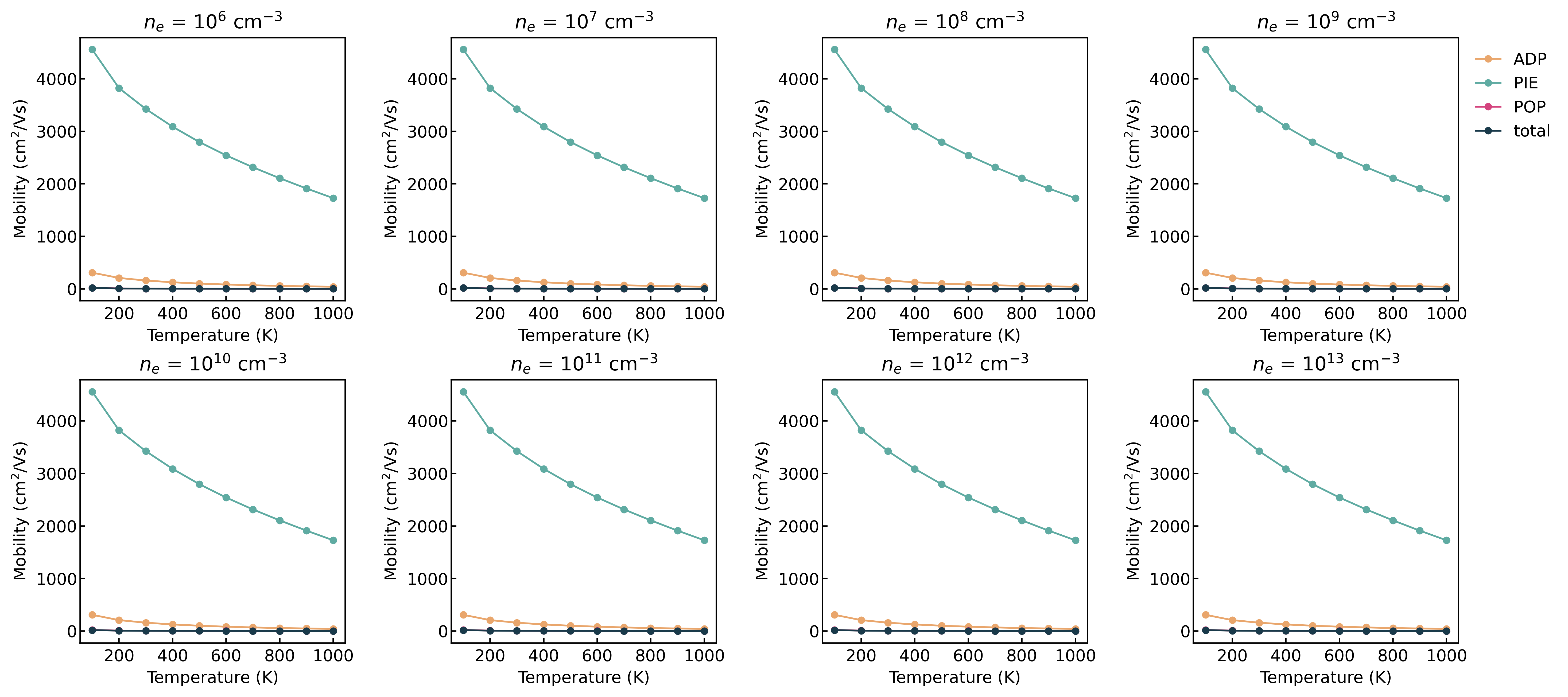
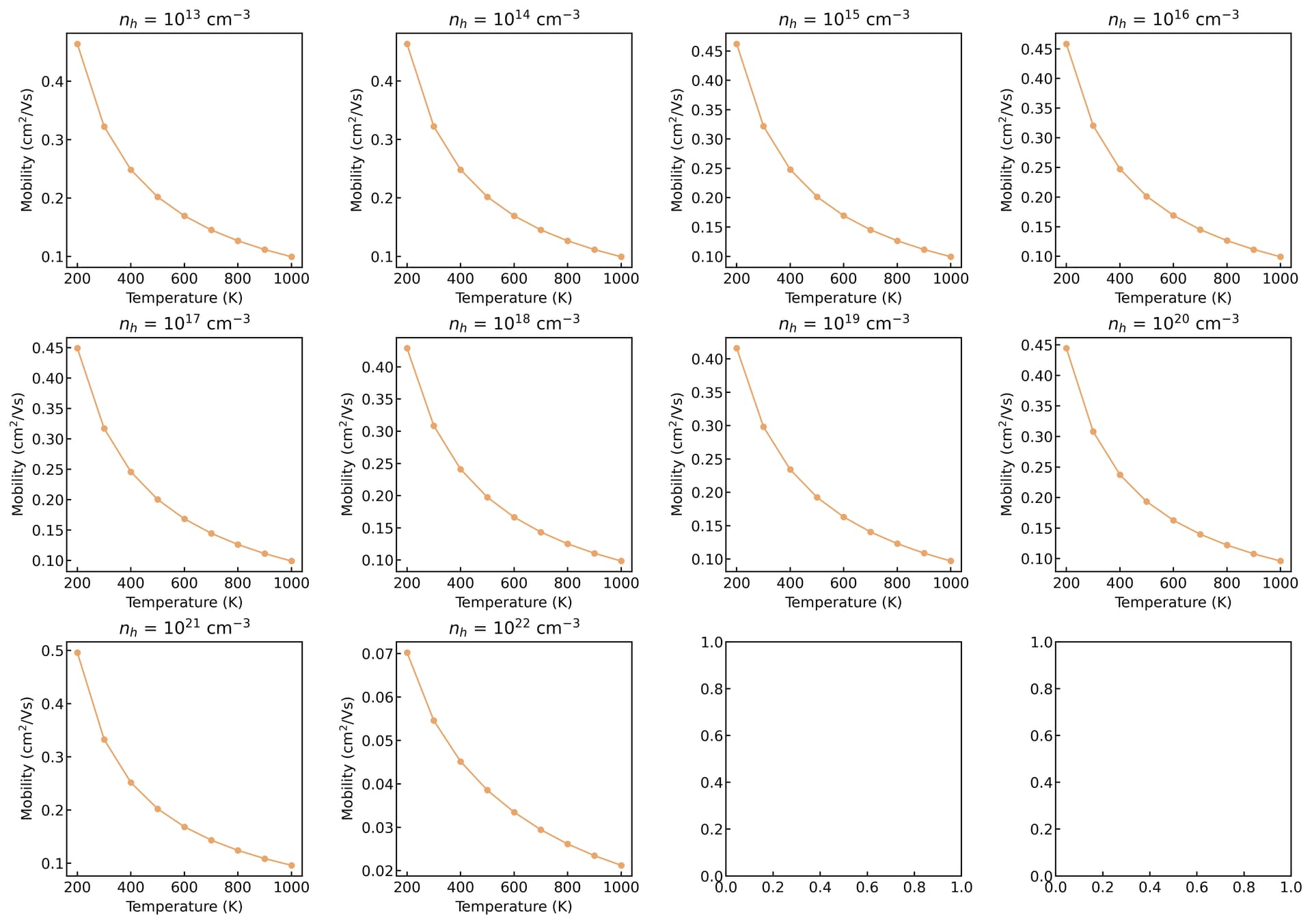
Irrespective of doping and temperature, I get the same mobility as shown in the attached Fig. I am a bit confused here since the amset run completes with a success message and I was able to use all the functionality of the amset to plot.
I searched all posts since 2020, but I don’t see any report on such an issue. Hence I use the tag “bug-report”. But I am not sure if is it a bug or wrong settings in the YAML file or i set an unsuitable scattering mechanism.
For better understanding, I attached the amset.log and setting.yaml with this message.
Has anyone faced a similar issue?
Your comments are highly appreciated
The mobility remains unchanged with doping concentration, likely due to the extremely low doping level in your calculation. This results in negligible changes to the position of the Fermi level (please verify this by plotting the Fermi level as a function of doping concentration).
But it does seem odd that the scattering rate remains completely unchanged, maybe the material has a relatively flat band maxima?
The only factor that might change significantly is the IMP scattering rate, but this was not included in your calculation.
Hi @Zane
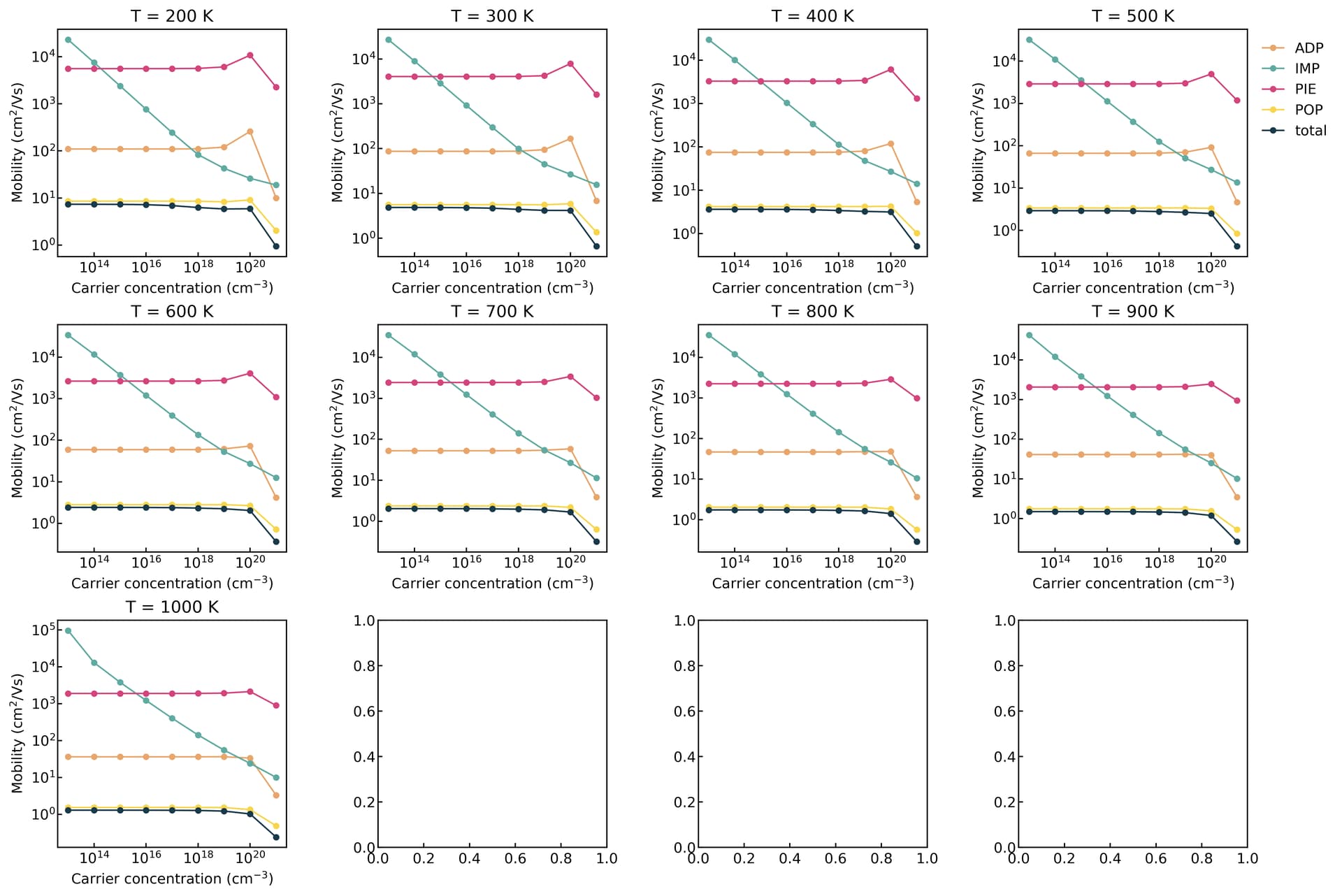
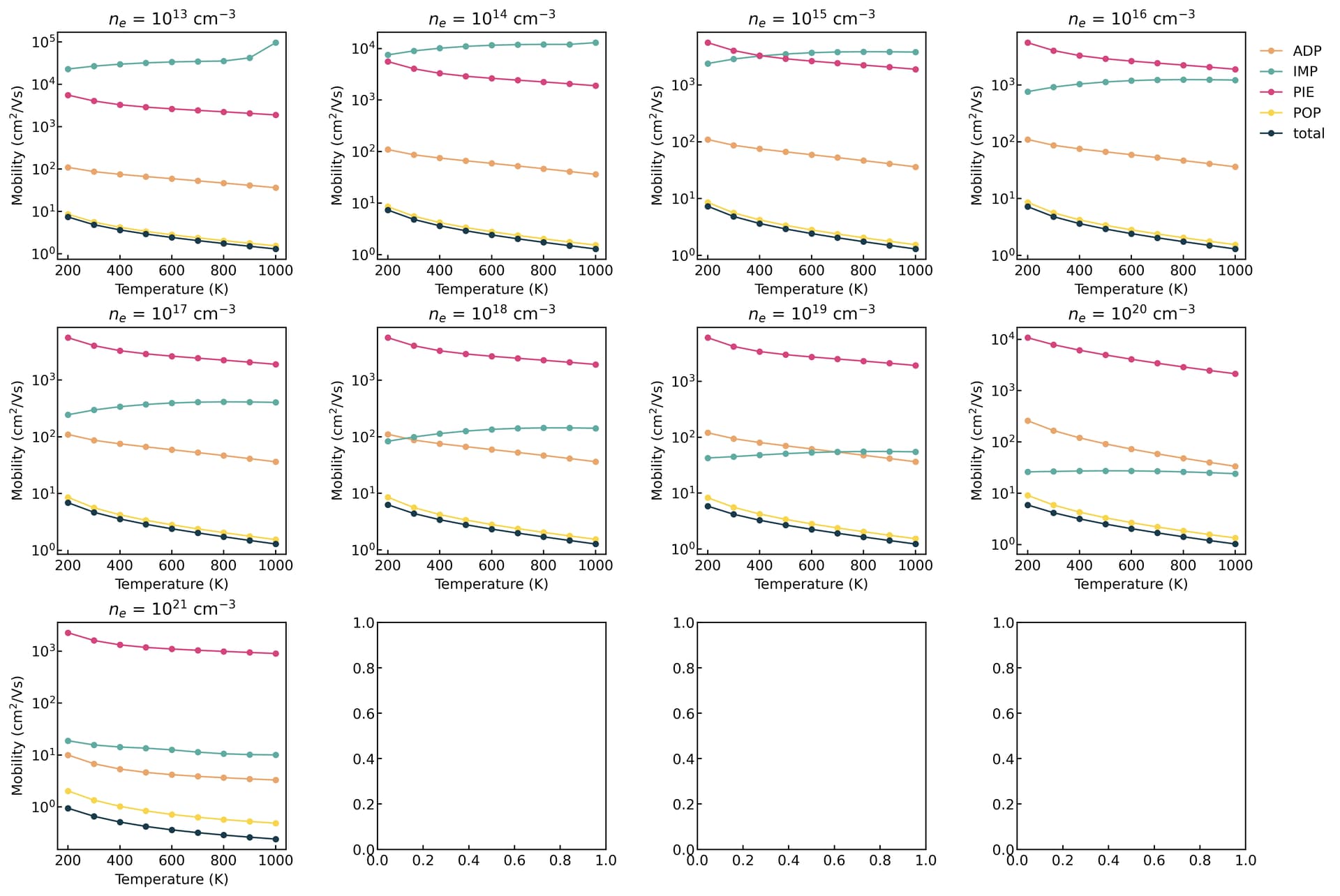
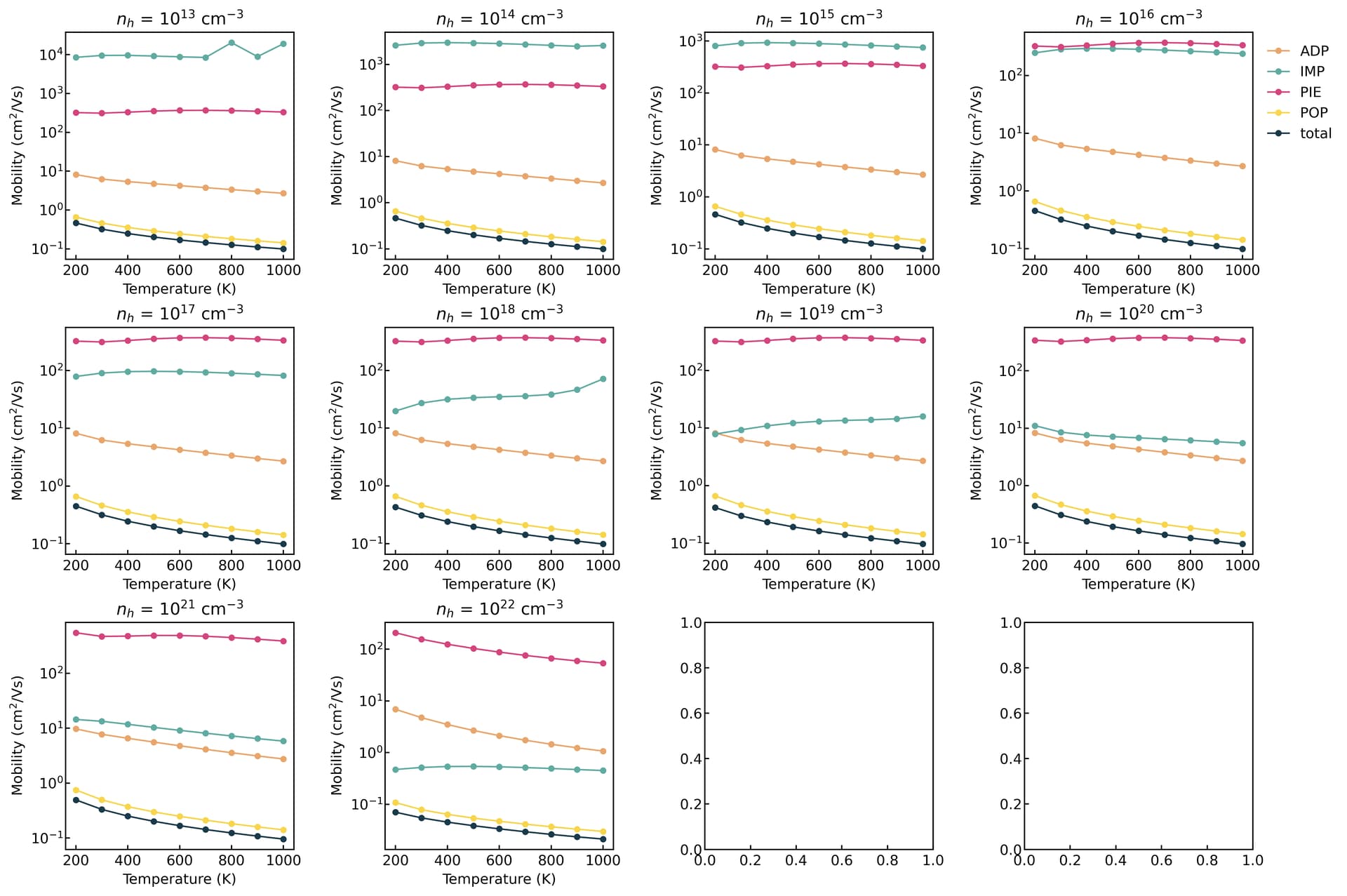
thank you for your interest and time on this issue. I agree that the doping level is low and no IMP is involved. so I did a separate calculation for this same material and the result is
(in terms of carrier concentration)
Are you suggesting that the conductivity at the CBM aligns better with experimental results than at the VBM?
Based on the diagrams, the carrier transport is dominated by POP scattering. If you do not turn on the free carrier screening, it typically remains insensitive to variations in carrier concentration.
thank you @Zane for your suggestions. finally, I see some variation. The strucutre I try is so far only a theoretical prediction. No experiment is available. again thanks for mentioning the free_carrier_screening option. setting it to true gives different results.
Moreover, recently opened other topics
I am just trying to control the number of bands involved in the whole calculation. Can you comment on it, if you faced such an issue
I believe the issue occurs because the band index from wavefunction.h5 does not align with deformation.h5.
In rare cases, even when using the same cutoff energy to extract the deformation potential and overlap matrix, the band indices may still be mismatched.
From my experience, the most reliable approach is to first extract the overlap matrix using a reasonable cutoff energy and then use the corresponding band indices to generate the deformation.h5 file.
In your case:
When generating deformation.h5, try: amset deform -b 13:16 …
thank you @Zane for your quick response.
I tried in many different ways to control the bands. But still I get the same error.
I created deformation.h5 with -b 13:16, -b 13:16 -e 200 and -e 200. If I do not use the -b option, things will go well with the automatically selected bands.
With the amset wave, the energy cutoff remains 200 eV for any combination of -b, which is bit weird.
in your answer i see
> I believe the issue occurs because the band index from wavefunction.h5 does not align with deformation.h5.
Then, how to confirm that the bands index is taken in the correct order?
if amset defor read and amset wave mix the band order, then does the result calculated by amset is reliable?