Hello,
I have ran into a problem while running with a custom potential. When I run a simple case (3 atoms, initial velocities ~ 10K, NVE), with any thermo output frequency > 1, the total energy (TE) of my system is not conserved.
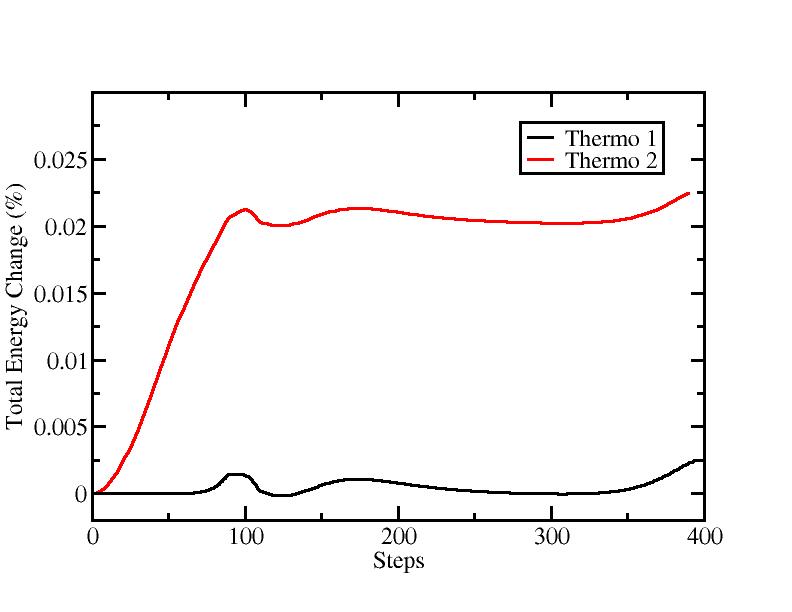
Specifically, the short time during which the initial velocities are redistributed shows strong change in TE.
But, when I set the thermo output frequency to 1, everything is fine.
Varying the dump frequency does not affect the results.
It really seems like writing the output file at every step is preventing the total energy from drifting. I am right now digging the code to understand what could go wrong, and what happens exactly when the thermo command is called. But I would like to ask potential and Lammps developers in if they ever ran into a similar problem or have an idea of its origin.
I am attaching a plot of the total energy after a few hundreds steps, as well as my input files. Unfortunately, the potential is not ready for release yet. It was written on a 2008 version of Lammps (where it ran without a problem) and I ported it to the 9 Nov 2011 version.
I would welcome any suggestions while looking for the issue.
Thank you,
Romain
in.3atom-1 (536 Bytes)
in.3atom-2 (536 Bytes)
3atom.input (227 Bytes)