Hello everyone,
In the past years I have used lammps to perform some simulations on coarse grained model, so I’m not a complete noob - though I’m no expert.
I was wondering if there was a “simple” way to define non symmetric pair potentials. I have in mind something like this: we consider 2 atom types. The pair interaction is e.g. lj for 1-1 and 2-2 interactions. But is like lj with some parameters for 1-2 and different parameters, or 0 for 2-1 (e.g. the first particle is repelled or attracted by the second, but the second feels a different force - violating Newton’s principle). I understand that simply using pair_coeff in the form 2 1 does not work. Do you have any suggestions? Thank you for your time,
Alessandro
1 Like
I have a rather fundamental question: If you have a pair of two atoms and one is of type 1 and the other of type 2, how will you be able to tell whether it is a 1-2 or 2-1 pair?
Violating Newton’s third law will require writing a custom pair style in C++.
You will basically have to implement a LJ pair style that uses a full neighbor list instedd of the (default) half neighbor list. As you already noted, you will also have to be “creative” in how you enter potential parameters, you may have to read them from a file like it is done with manybody potentials.
I don’t even want to go into the specifics of what violating Newton’s third law means to the thermodynamics and statistical mechanics of the simulated system…
The most straightforward way to achieve this is to write a custom fix, which can request a neighbor list and iterate over it during post_force to calculate and add on any force you like, no matter how gloriously non-physical. I am deeply skeptical it is a good idea (how will you conserve momentum or energy or use a thermostat) but if I had to do it, that’s how I would.
Thank you very much for your feedback.
Indeed I was thinking of “hijacking” lammps to perform a simulation of a dynamical system with no real thermodynamics backing it up, à la “particle life”.
See e.g.:

I was hoping to be able to use lammps because it has a very convenient scripting language and is very fast - instead of having to write most code from scratch. The idea of using a homebrew version of lj potential is good, though being a lazybum I hoped I could get away with some dangerous/unlawful scripting hack ![]()
Thanks again,
ALS
I have found a full neighbor list version of pair style lj/cut called lj/cut/full, that I had written some time ago for testing purposes, lying around on my hard drive.
I am attaching it here. So to implement your model, you would just need to “massage” the code in the coeff() and init_one() functions to remove the symmetrization of the LJ parameters and disable the reordering in the Input::pair_coeff() function in input.cpp
or set one_coeff = 1; in the pair style constructor and write a reader for all parameters from a file in one go, like it is done for many-body potentials like tersoff or sw.
pair_lj_cut_full.h (1.3 KB)
pair_lj_cut_full.cpp (7.2 KB)
3 Likes
Thank you very much ![]()
I believe this can be done in vanilla LAMMPS, albeit it is quite hacky. It requires using multiple fix nve, separately for each atom type, and pair_style lj/cut/coul/cut. You alternate between nve fixes so only one is active during a single step. Between steps, you change the charge of a chosen type of atoms. You also need fix viscous as this setup constantly creates kinetic energy, see the animation below:
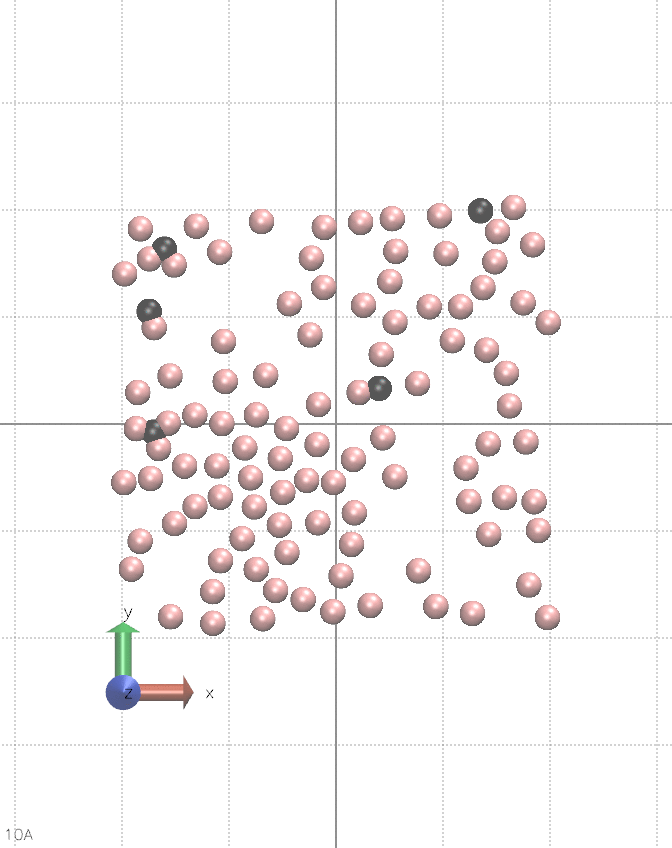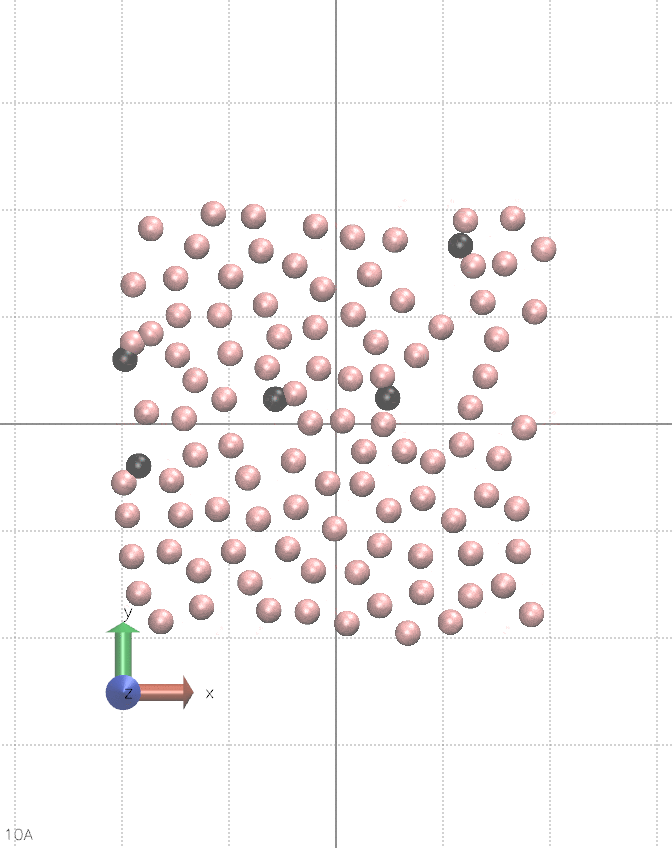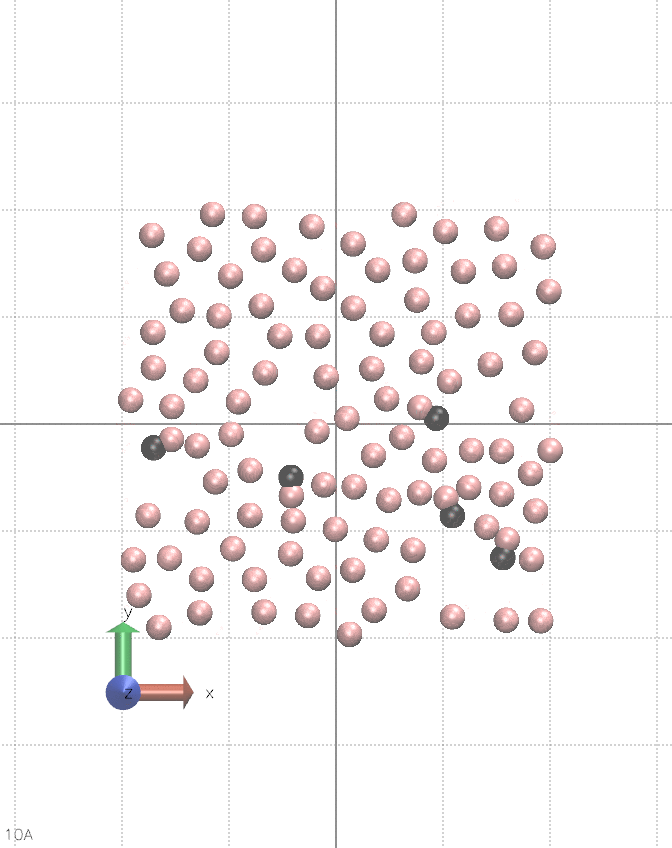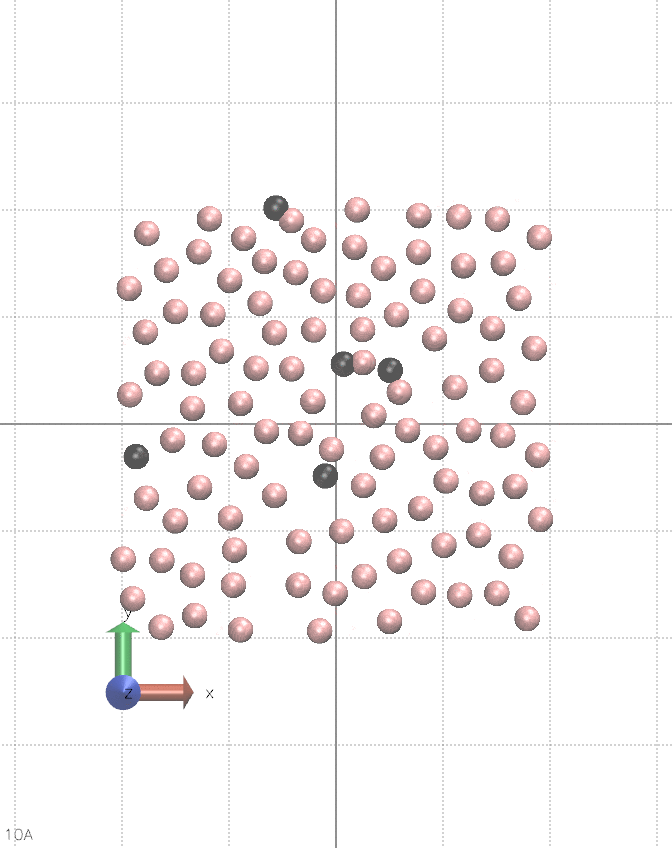
The input script for the animation is below.
dimension 2
units real
atom_style charge
region u block -20 20 -20 20 -0.5 0.5
create_box 2 u
mass * 100
create_atoms 1 random 5 144 u
create_atoms 2 random 100 14234 u
pair_style lj/cut/coul/cut 6.0
pair_coeff * * 1 2
group 1 type 1
group 2 type 2
set group 1 charge 2
set group 2 charge -2
minimize 1e-10 1e-10 10000 10000
dump dump all custom 10 dump.lammpstrj id element x y z
dump_modify dump element Co Fe
thermo_style custom step pe ke temp cpuremain
thermo 100
run 0
timestep 0.5
fix viscous all viscous 10000.0
variable loopa loop 20000
label loopa
fix nve1 1 nve
run 1
unfix nve1
set group 2 charge 2
fix nve2 2 nve
run 1
set group 2 charge -2
unfix nve2
next loopa
jump SELF loopa
2 Likes
Thanks, I’ll check this too