I am running an NPT simulation with some surfactant molecules, water molecules and rigid MoS2 sheet.
After some steps water molecules keep on building along z-direction with simulation box shrinking along x and y direction:
Step TotEng E_vdwl E_coul Lx Ly Lz Temp
100000 -172120.39 57514.723 383598.89 150 150 150 302.39927
110000 -174091.31 58790.565 379812.08 149.33445 148.94266 148.45191 301.28535
120000 -175353.54 59036.393 378354.4 147.44846 147.84024 146.61407 298.71191
…
320000 -189607.81 64052.626 354556.44 89.374474 88.258761 95.857047 300.25466
330000 -189984.14 64396.35 353744.31 88.698804 87.609252 96.24154 298.76996
340000 -190382.53 64239.002 353098.06 88.091419 86.987032 96.295348 300.41513
…
770000 -193657.45 65824.09 346731.28 80.874996 82.685272 106.65229 299.86779
780000 -193638.16 65862.302 346673.79 80.811633 82.569225 106.91442 299.82986
790000 -193677.01 65874.631 347042.42 80.42274 82.651359 107.02641 298.50931
…
2430000 -194013.54 65719.867 346952.29 69.44792 68.568661 149.78615 298.56992
2440000 -194037.91 65981.87 346124.8 69.384034 68.727333 149.79766 300.59061
2450000 -193869.81 66135.617 346225.46 69.044234 68.77927 150.11355 299.60018
ERROR on proc 58: Out of range atoms - cannot compute PPPM (…/pppm.cpp:1918)
Application 685460 exit codes: 134
Application 685460 exit signals: Killed
Application 685460 resources: utime ~53997s, stime ~462s, Rss ~42612, inblocks ~738252, outblocks ~2307784
Is there any known reason for such behavior? This only happens when i add rigid MoS2 sheet in simulation (i.e water-surfactant system runs perfectly fine in same conditions).
Lammps 10Aug2015, Coefficients assigned through moltemplate.
formatted input file and output file cane be accessed here:
Input file: http://pastebin.com/KUMzjMnM
output file: http://pastebin.com/ttwHPr4s
Your fix npt command must be allowiing that to happen,
i.e. different dimensions to grow/shrink independently.
And it would be driven by the Pxx, Pyy, Pzz components
of the pressure tensor. So if you monitor those in thermo
output I imagine you will see they are consistently negative
or positive, which will shrink/grow the corresponding dimensions.
Steve
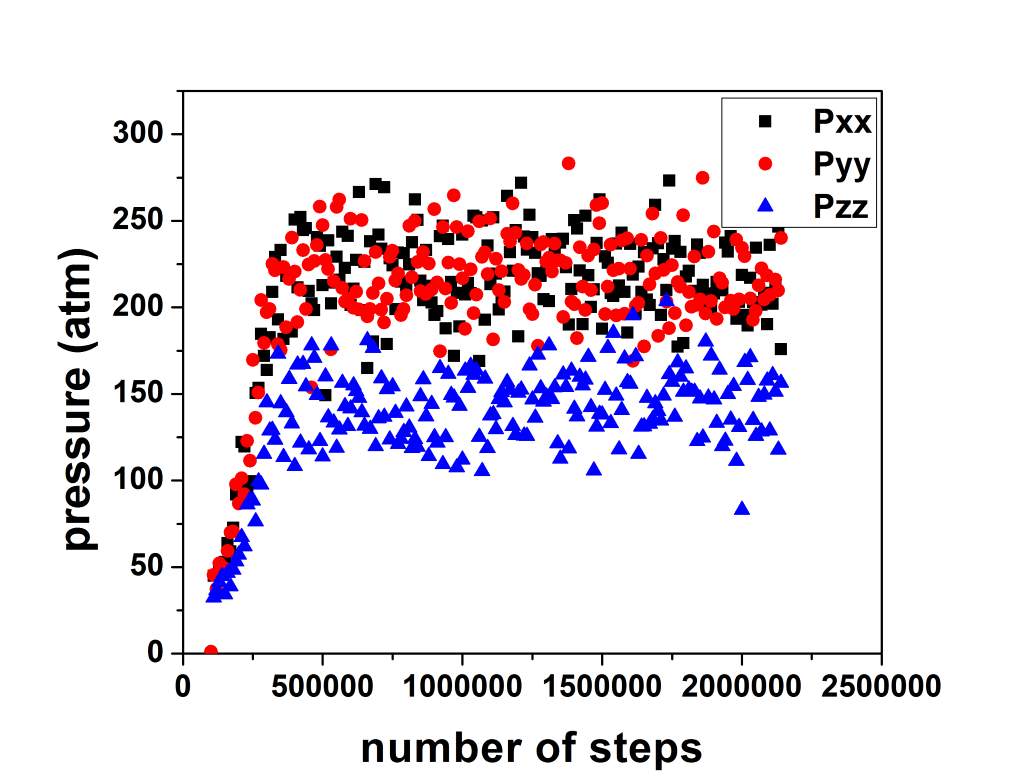
Thank you for reply. I ran simulations again with thermo_custom set to give out pxx, pyy, pzz. Indeed as you suggested pzz is nearly half of pxx and pyy. Also values for all of them is really high [Graph1]. If i couple them with following line:
fix NPT MovingAtoms npt temp 300 300 100 iso 1.013 1.013 100 drag 1
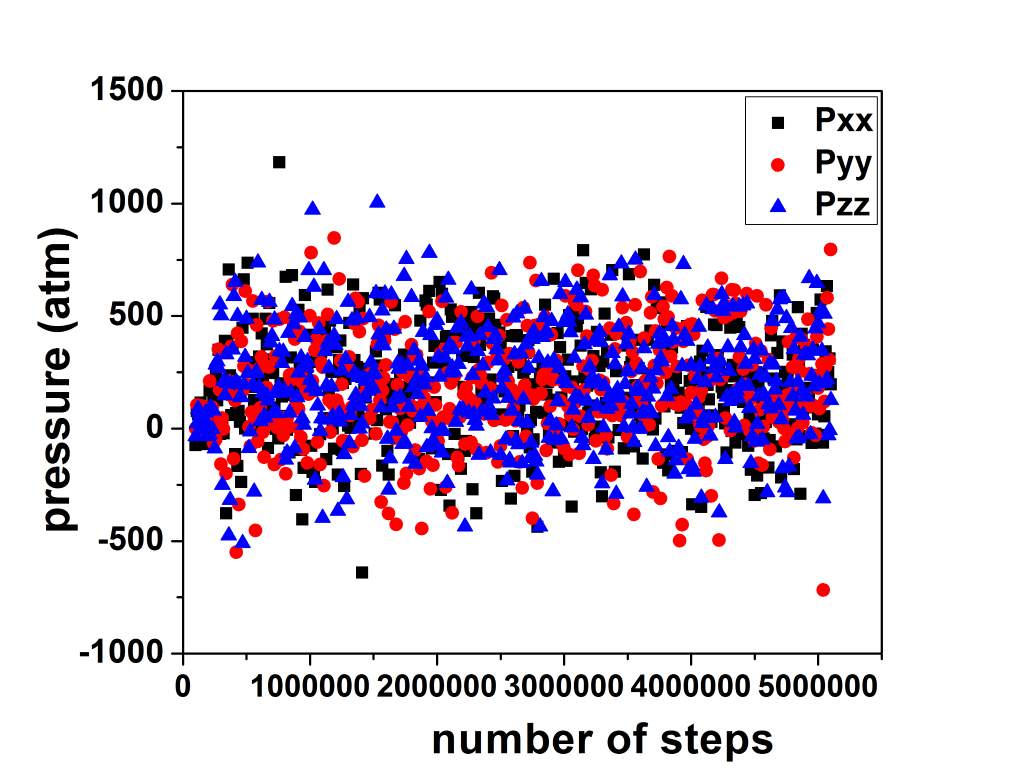
There is no stretching in Z direction but Pxx, Pyy Pzz fluctuate a lot [Graph 2]. I understand that it is due to damping factor being 100 so testing now with increasing values of damping coefficient.
I have 3 more questions however:
-
Is there any particular reason for Pxx, Pyy and Pzz to be this high in graph 1? also any reason for pzz being so low?
-
Error I got was out of range atoms, as far as i can understand from previous mails in mailing list, that generally happens when atoms move so fast that they travel more than half the dimension of box. But here it is a gradual increase in one direction. Shall LAMMPS not accommodate it?
-
What happens when box tries become smaller than the dimensions of a rigid body?
Thank you.
Hi.
Incidentally, I tried running the system that Amit posted on 1/26, and
there were definitely some problems with the physics. The system
exploded. (I was also able to get LAMMPS to segfault.) I'll post
these details and input-files in the other thread which discusses the
segfault. I suspect this is related to the problems you are
discussing in this thread.
There is no substitute for looking at your system visually.
Amit, do you know how to do this? If you look at how the atoms move,
you will be able to see something is very wrong. One way is to use
VMD. If you have VMD installed, then when you run moltemplate this
way:
moltemplate.sh -vmd system.lt
... it creates a file ("system.psf"). Later, after your simulation
has finished, you can run VMD this way:
vmd system.psf
In the white box in the "VMD Main" window, right-click on the
"system.psf", and select "Load Data Into Molecule". Then click on the
"Browse" button, select the dump file you created earlier, and select
"LAMMPS Trajectory" from the "Determine File Type" pull-down menu.
Then click "Load".
Cheers
Andrew
Please forgive me Amit.
I did not intend to reply to your post but I got your name confused
with a different moltemplate user named Ankit.
I'm embarrassed about this. Forgive me.
It seemed like Ankit had not looked at his simulation visually yet, so
I suggested that's what he do. That advice certainly applies to you,
if for some reason, you have not yet looked at your simulation
trajectory either. It sounds like you are using moltemplate, so the
advice I gave (using the "-vmd" option) should work. However if
you're not using moltemplate to build your system, I attached some
more general instructions to this message. (Just remember to change
the name of the data file from "system.data" to the name you gave your
data file.)
Other random advice:
It is often helpful to simplify the system, one step at a time until
the problem goes away.
For example: it is tricky to run NPT simulations when a large sheet is present.
(It sounds like you are using fix npt with the "aniso" keyword. That's good.)
Does the system explode if you remove the sheet from your simulations?
Alternatley, have you tried removing the surfactants?
(As you probably know, if you are using moltemplate, you can do this
by commenting out the line(s) where you instantiate these molecules.)
Good luck
Andrew
README_visualize.txt (2.85 KB)
Sorry for the delayed reply. Yes I do visualize the system, but with simple lammpstrj file, i.e. dumping data periodically to lammpstrj file and loading it in vmd followed by unwrapping (pbc unwrap). So far (as stated in my previous mail), i only managed to get a stable system after giving “iso” keyword in npt fix. But pxx, pyy, pzz fluctuate a lot. Also as i stated it need not necessarily explode (although it certainly did initially, i was taking it as a feature of large vacuum chunks present in my initial simulation box which accelerate molecules to high speeds under npt, resulting in atoms lost error), as in my case it gradually shrinks in x and y direction. Energy is stable, dimensions grow slowly. That is why i wanted to know in previous mail:
“”"
-
Is there any particular reason for Pxx, Pyy and Pzz to be this high in graph 1? also any reason for pzz being so low?
-
Error I got was out of range atoms, as far as i can understand from previous mails in mailing list, that generally happens when atoms move so fast that they travel more than half the dimension of box. But here it is a gradual increase in one direction. Shall LAMMPS not accommodate it?
-
What happens when box tries become smaller than the dimensions of a rigid body?
“”"
As i was thinking that problem is due to boxes shrinking more that dimensions of rigid sheets, which results in some sort of overlap of atoms. But even then most likely error should be neigbouring list overflow and shall i not see a sharp jump in energy?
Till now i have played with drag, pchain keywords but no avail, i.e. giving iso prevented any crash but pressure fluctuates a lot.
Apologies! Clicked send little early! When I do remove the sheet (i.e only water and surfactant in simulation box) it does not explode or crash. However pressure tensor components still fluctuate, though spread is very less (I did attach graph of it previously). I will try just water and sheet, but it will take a litle while as all ques are full right now at local cluster.