Dear LAMMPS users,
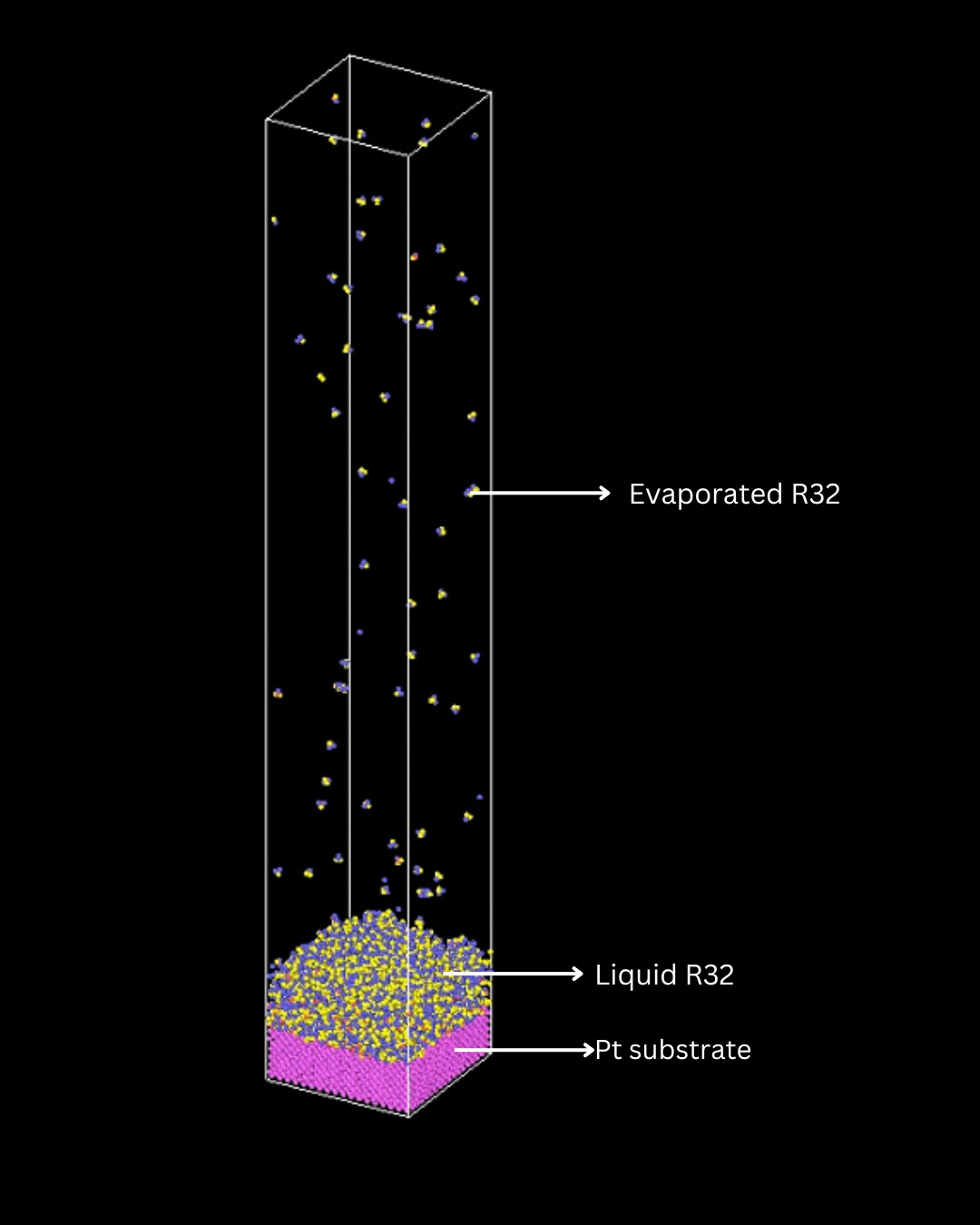
I am currently simulating the evaporation of R32 (CH2F2) refrigerant molecules on a Platinum surface. In an output dump file for each time step, I need to find out the number of evaporated R32 molecules.
For a previous simulation similar to my current one, I simulated the evaporation of argon atoms from the Platinum surface, and to find the number of evaporated argon atoms, I used the “coordination analysis” in OVITO, where a boolean expression "Coordination < 7 && Position.Z > 0 && ParticleType == 1’ gave me my number of evaporated atoms for each time step.
The boundary conditions were periodic in x and y directions and fixed in the z direction, and argon had Particle Type 1.
The boundary conditions are the same for my simulation with R32, but counting the number of evaporated R32 molecules with the same principle does not apply anymore as can be seen. Is there any way to obtain this using the information from dump files?