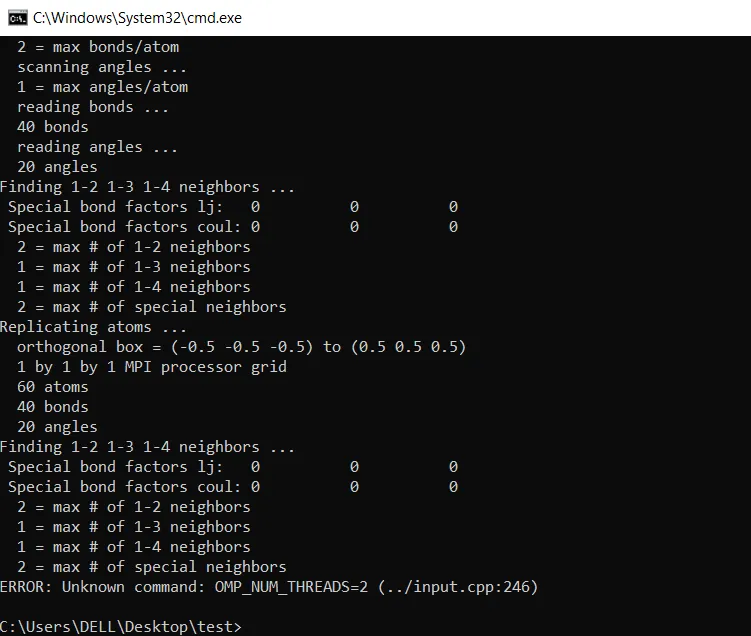
Error: Unknown command: OMP_NUM_Threads=4
Why this line is shown? please help
Error: Unknown command: OMP_NUM_Threads=4
Why this line is shown? please help
Not enough context.
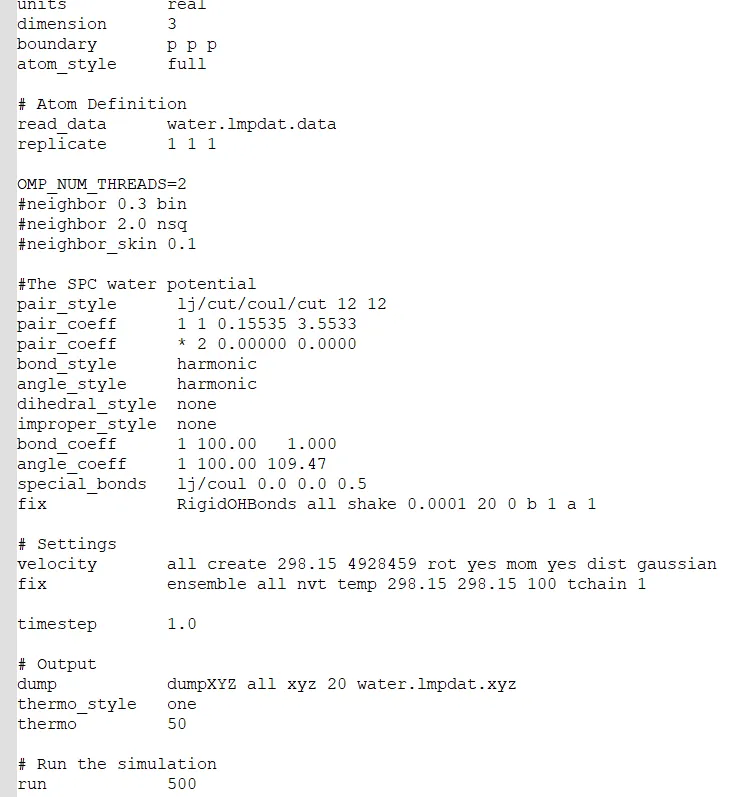
Here is my code for simple few water molecules.
Another problem I am facing i.e. “proc sub-domain size < neighbor skin, could lead to lost atoms”
Please help!
Where in the LAMMPS manual does it say that OMP_NUM_THREADS=2 is a LAMMPS command?
The error message is well deserved.
Look at your output! It says you have a 1x1x1 Angstrom sized box. Does that make sense to you?
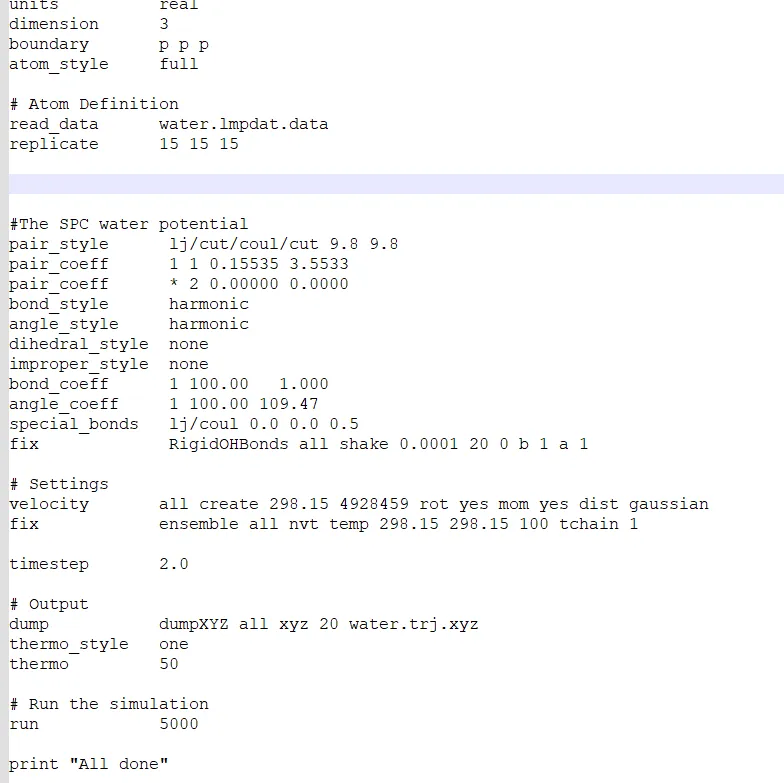
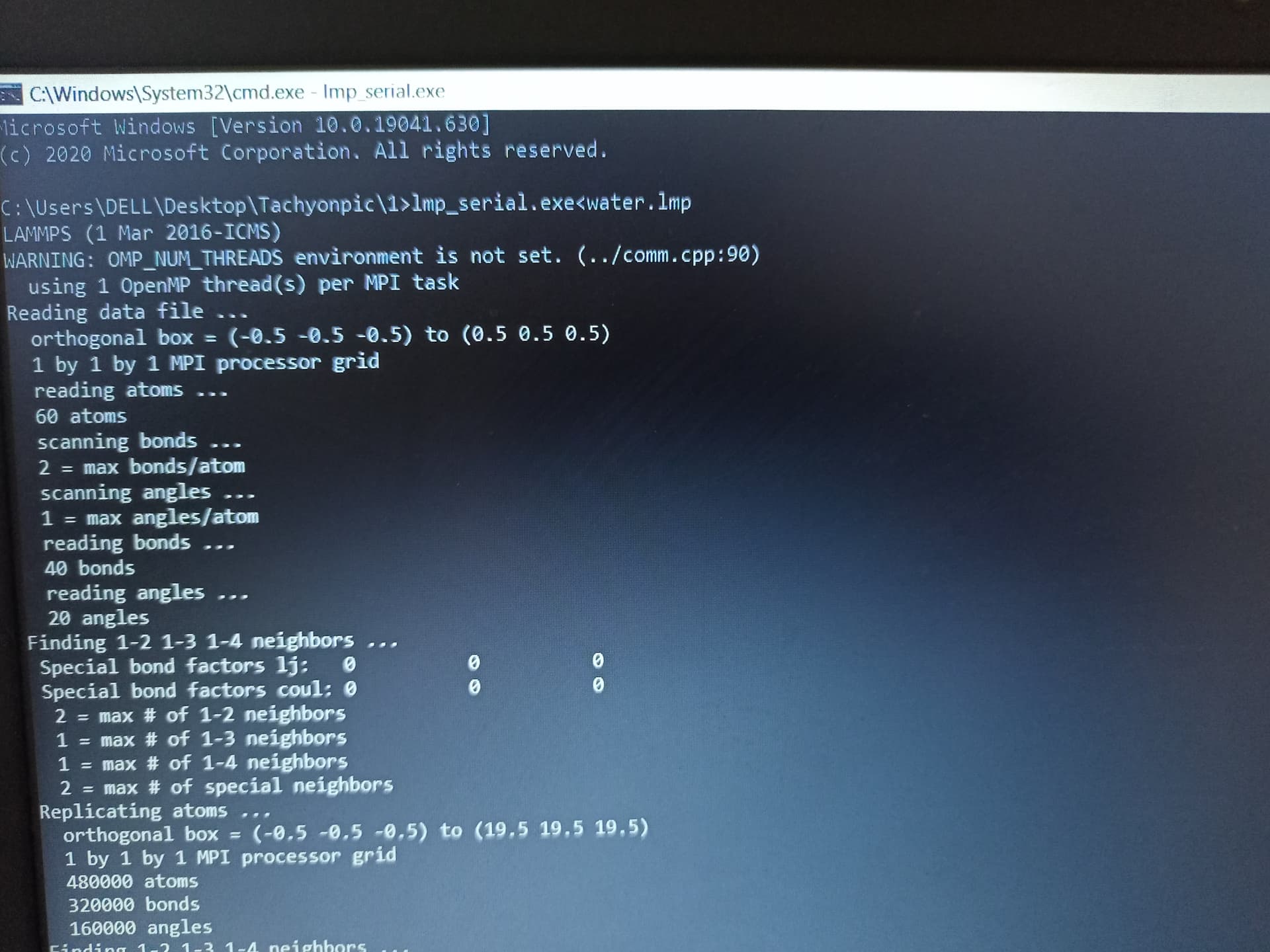
Yes! I am doing wrong in this case. However, I write the box to 15×15×15. When run the input file, I find output file without any data. Why it is happens?
The information you provide is insufficient to say anything.
Also, using the replicate command does not correct your incorrect box dimension. You probably just ran out of memory and there was an error you did not show.
That warning is harmless.
But your Screenshot confirms that you are still using the bogus data file. Also your are using an obsolete version of LAMMPS.
You need training and guidance at a very basic level about how to run software, how to study and apply what is written in a software’s documentation, how to confirm that what you are using as input is meaningful and much more.
This is way beyond what can be taught from remote and via this forum in particular.