Hello everyone ![]()
I am using this post to carry out a participated peer review of a paper, which I hope to publish in the next few months. It is about a coarse-grained force field for LAMMPS based on ellipsoids decorated with point charges and connected by directional bonds. Its main features are:
- It reproduces the excluded volume of molecules ( → overlapping ellipsoids).
- It includes long-range electrostatics, yielding good condensed-phase properties.
- It can be back-mapped to atomistic coordinates without losing structural information (as the CG beads are finite-size particles, they also have a quaternion describing their orientation).
The first implementation was based on lammps-30Oct19 as a hybrid pair_style. Here is the accompanying paper: MOLC. A reversible coarse-grained approach using anisotropic beads for the modelling of organic functional materials (2019).
I left academia after publishing this paper, and these days I am a scientific consultant. However, I keep working on the MOLC model as I am pretty confident about the quality of the resulting simulations. I am preparing a paper describing a new parameterisation strategy, plus bits which are of general interest to the MD community (e.g. a new compute for the intermolecular energy of force-fields with long-range electrostatics).
Meanwhile, the MOLC model has been ported to lammps-2Jun22 and transformed into a pair_style, which is a combination of a Gay-Berne potential plus classic electrostatics with point charges. The original plan was to submit the new code to the main LAMMPS repo after peer review, but the developers would need to audit the code anyway. So I asked directly one of them, as it’s unlike that a referee will have the time and resources to scrutinise the code and verify the consistency of the physical equations.
Let’s take the chance to run a little experiment together. I will discuss the paper’s open issues and technical details in this post. I have asked @akohlmey to referee the paper before submission, and I will include him as a co-author to acknowledge his precious contribution. I have received countless suggestions in this forum, and it is clear that erroneous results can and indeed do pass peer review, especially for very technical subjects. I recently read an interesting post questioning the efficacy of peer review, so let’s try an open review instead. Feel free to share your thoughts on this approach or the scientific aspects.
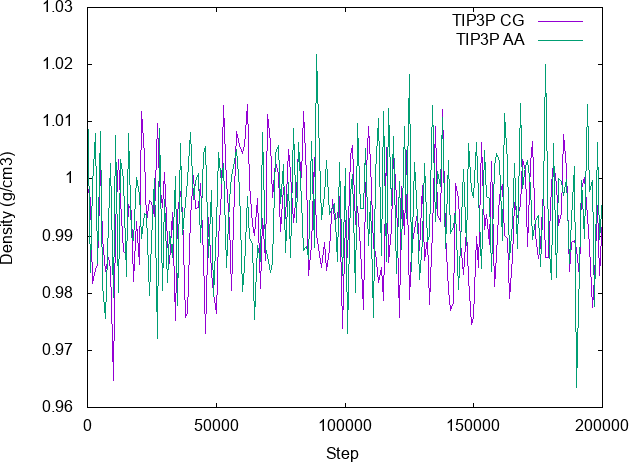
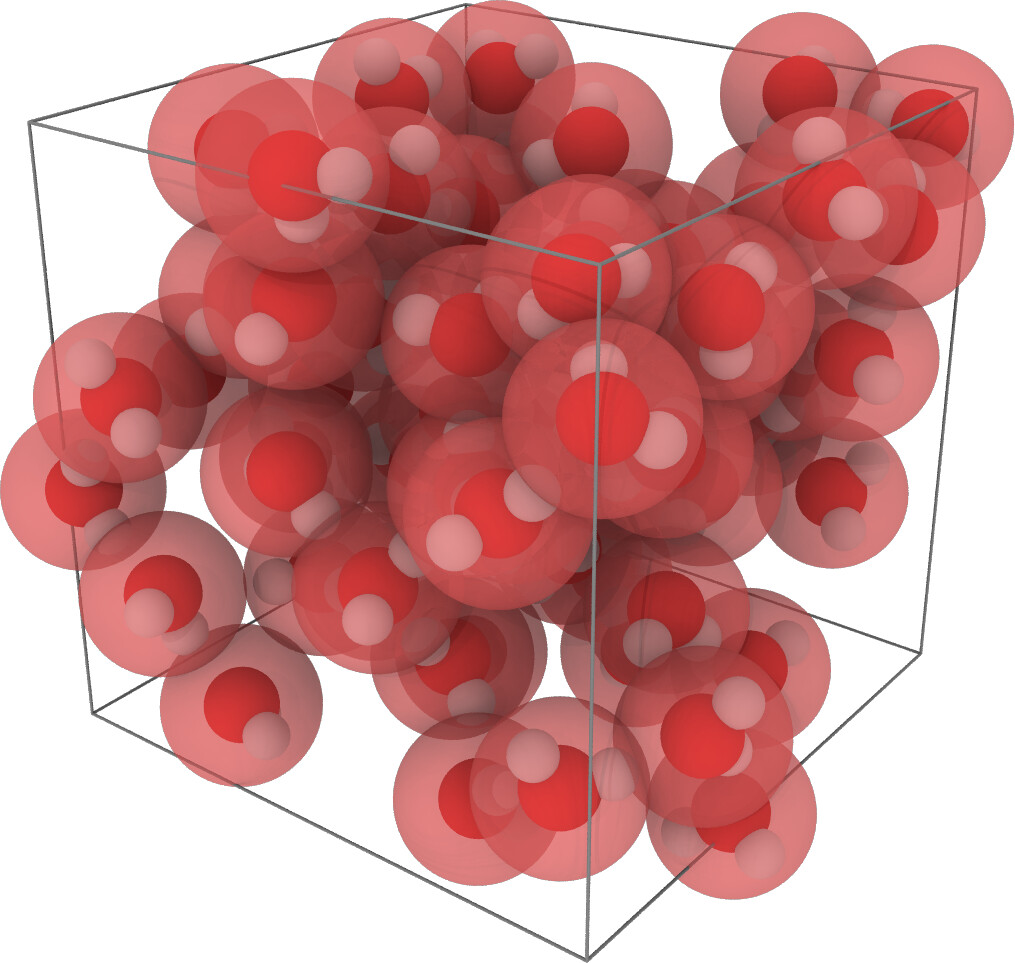
The paper deals with the parametrisation of linear alkanes. Here is a teaser showing the CG model of n-hexadecane (C16H34) superimposed to the back mapped atomic coordinates: 
I will post anything specific to the MOLC model here and discuss more general issues on the #lammps forum. Keep you posted!