I am trying to use a conditional to set the pair_style and kspace_style depending on whether the system contains finite charges or not BEFORE a run. My idea was to take the max and min of the ‘q’ atom vector, and write a conditional based on those. However, I cannot figure out how to do this or if it’s possible. The issue seems to be that the special functions ‘max’ and ‘min’ can only operate on global vectors, not atom vectors. I can get the result if I use ‘compute reduce’, but that only gets evaluated after a run. Here is a mockup of what I’m trying to do (omitting the min for simplicity), based on the 8.4.3. TIP3P water model — LAMMPS documentation example:
units real
atom_style full
region box block -5 5 -5 5 -5 5
create_box 2 box bond/types 1 angle/types 1 &
extra/bond/per/atom 2 extra/angle/per/atom 1 extra/special/per/atom 2
mass 1 15.9994
mass 2 1.008
bond_style zero
bond_coeff 1 0.9574
angle_style zero
angle_coeff 1 104.52
molecule water tip3p.mol
create_atoms 0 random 33 34564 NULL mol water 25367 overlap 1.33
variable qatom atom q
if “$(max(v_qatom))>1.0e-6” then “pair_style lj/cut/coul/long 8.0” “kspace_style pppm 1.e-6” &
else “pair_style lj/cut 8.0”
This does not work because of the mismatch not allowing max() to work on atom vectors. I also seemingly cannot make “qatom” a vector-style variable, because it won’t properly assign q to it (I will get a zero length error). Is it possible to do what I’m trying to do here, and if so, how?
For as long as you use pair/bond/angle/dihedral/improper styles zero, there is no harm done with issuing a run 0 post no
With the latest development version of LAMMPS, the following is possible.
atom_style full
region box block -5 5 -5 5 -5 5
create_box 2 box bond/types 1 angle/types 1 &
extra/bond/per/atom 2 extra/angle/per/atom 1 extra/special/per/atom 2
mass 1 15.9994
mass 2 1.008
bond_style zero
bond_coeff 1 0.9574
angle_style zero
angle_coeff 1 104.52
run 0 post no
molecule water tip3p.mol
create_atoms 0 random 33 34564 NULL mol water 25367 overlap 1.33
compute q all property/atom q
compute min all reduce min c_q
compute max all reduce max c_q
print "min: $(c_min) max: $(c_max)"
Based on that information you can then change pair style and kspace style and so on to whatever you like.
Please also note that to do this safely, you may also first need t check via the is_available() variable function whether atom style and kspace style (and any other styles) are actually available for use or using them would create an error.
The end goal for me is to encapsulate these commands inside an OpenKIM Simulator model (we are adding large bonded force fields that support a variety of charged and neutral atom types, so we don’t know ahead of time if the user might choose to run with all-neutral atoms). I’m a little hesitant to use your solution because right now the user is allowed to hold off on setting the masses until after calling “kim interactions”, in which case a “run 0” wouldn’t work. Do you see any issues with the following option?
pair_style lj/cut 8.0
label loop
variable i loop $(atoms)
if “$(q[v_i])!=0.0” then “pair_style lj/cut/coul/long 8.0” “kspace_style pppm 1.e-6”
next i
jump SELF loop
I think we specifically want to omit is_avaliable(), because we want LAMMPS to exit if it was not built with the styles we expect, and the default LAMMPS error messages are quite sufficient.
EDIT: I see that “jump SELF” may be less than universal depending on how the input is run, so this may not be the perfect solution
I don’t understand the problem you are trying to solve. If you want to set the force field via OpenKIM, it is the model that has to determine what the (partial) charges are. Proper atom typing and assigning the corresponding charges for molecular force fields is a highly non-trivial and complex task, so I don’t see how you can do that at all from just spewing out some “generic” LAMMPS input script code. Particularly, since this would have to be done differently for different force fields and in a situation where you know far less about the system at hand than what the force field specific tools know. Not to mention that those usually only provide a starting point and that it is often necessary to correct/update those (manually) after the fact.
You are setting a dummy force field, why not set dummy masses, too? You are already so far down the rabbit hole, a little further does not make a difference.
I disagree on that one, too. You do not want (possibly cryptic) error messages to happen from some input code that the user doesn’t know about and thus can be easily confused (“I never issues that command!”). Instead, introspection is the way to go. That we you can check ahead whether a model can be applied at all and then refuse early on with a proper error message from the KIM commands.
I think using the water example may have made things confusing, because it’s not representative of what we are actually trying to do.
The in-development support for bonded force fields as KIM LAMMPS Simulator Models works as follows:
-The simulator model encapsulates some LAMMPS commands to set up the pair_style, bond_style, etc., analogous to current LAMMPS simulator models (so I am not talking about a “generic” LAMMPS input script, I am talking about the set of commands contained in smspec.edn that is bundled with a specific force field)
-It also contains a file with coefficients for every pair, bond, angle, etc. supported by the force field
-The user must define their atoms and bonding topology using a labelmap. Depending on the force field, the user may also be expected to provide their own charges (for example, in IFF, the same atom types may have different charges depending on the environment. On the other hand, in something simple like core/shell models, the charges are always fixed and can be provided by the KIM model). The user does not provide any _style or _coeff, this is done in the next step
-After the atoms and bonding topology are created, the user issues “kim interactions”, which issues the aforementioned LAMMPS commands to set up the interactions (as it always has for Simulator Models), but also has new functionality that matches the type labels to the correct coefficients provided by the model
So the practical example is this. We have an KIM model for IFF. The force field is generally intended to be run with “pair_style lj/cut/coul/long”. However, the user may choose to use the model to simulate bulk Aluminum, in which case all the atoms are neutral and LAMMPS will raise an error. So I wanted to include a conditional in the KIM model definition that checks whether the simulation contains any charged atoms.
We already do something similar with Buckingham Simulator Models, where we check for periodicity (using a LAMMPS variable defined by the KIM LAMMPS package) to switch from coul/long to coul/cut. If there’s no good way to check for charge through LAMMPS commands, we will add the functionality to the KIM package.
Not sure if this is still relevant with my updated explanation (we are not trying to set a dummy force field), but the reason I wouldn’t want to set dummy masses is because if the user forgets to set their masses, we don’t want them to run dynamics without incident with the dummy masses we set.
This sounds sketchy to me. Partial charges are force field parameters, so either they should be fixed by the force field, or the force field in whole should be supplied by the user.
This seems reasonable to me, we will discuss this internally. Nevertheless, the original need to detect charges still stands even if all charges are prescribed by the KIM model. If the force field contains neutral atom types and the user chooses to include solely those, the issue still arises.
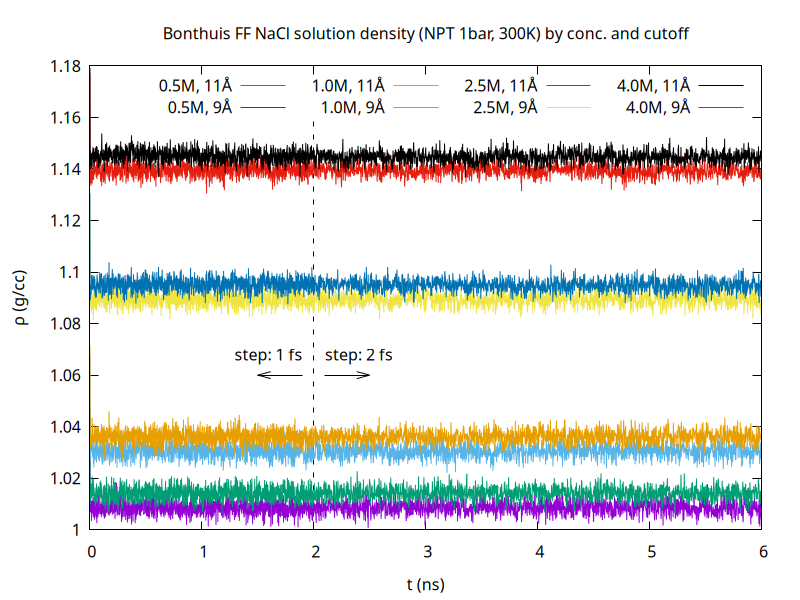
Not just charges – cutoffs and tapers, mixing rules, and long-range treatments are all part of the force field as well. For example, I recently tested a forcefield for NaCl in SPC/E water at four different molarities with NPT runs at 1 bar and 300 K. Simply changing the cutoff from 9 angstrom (the SPC/E default) to 11 angstrom (because I was subsequently going to combine this with some CHARMM-derived parameters) has an observable effect on the average density:
For another example, lots of biomolecular force fields will smoothly taper LJ forces between an inner and outer cutoff, so a user who wants to replicate those results must use an entirely different pair style from lj/cut/coul/long, such as lj/smooth or the charmm or gromacs variants.
So it seems to me that any “replicable packaging” for a force field has to select between many different options of pair_style and pair_modify anyway, acting on the script either extremely early in the initialisation process or from outside LAMMPS altogether.
Each force field would come with a bespoke specification of _style and _modify commands, and we are not averse to archiving different versions of the same force field. For example we are already planning to archive IFF with both 12-6 (lj/cut/…) and 9-6 (lj/class2/…) pair_styles as separate OpenKIM models. In fact, once we begin to develop OpenKIM Tests for these models, it will provide an automatic way to inspect precisely the types of differences you’re talking about (if we archived the force field you are using as two separate models with different cut-offs). This is not entirely different from different tabulations or spline types in reactive interatomic potentials, which we also sometimes archive as different models, although of course the number of possible combinations in bonded force fields is much higher, and we can’t hope to archive every possibility.