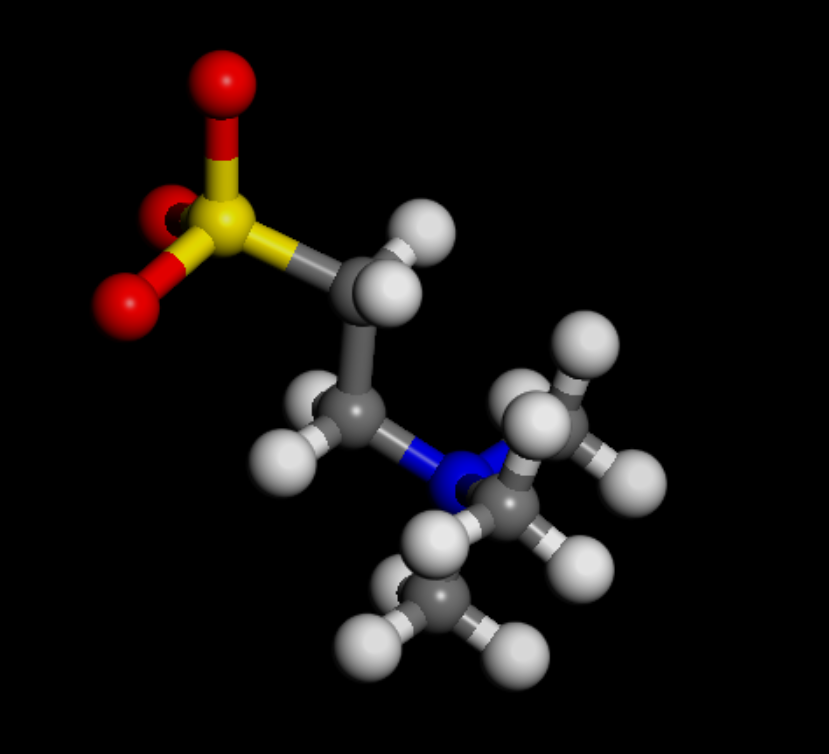
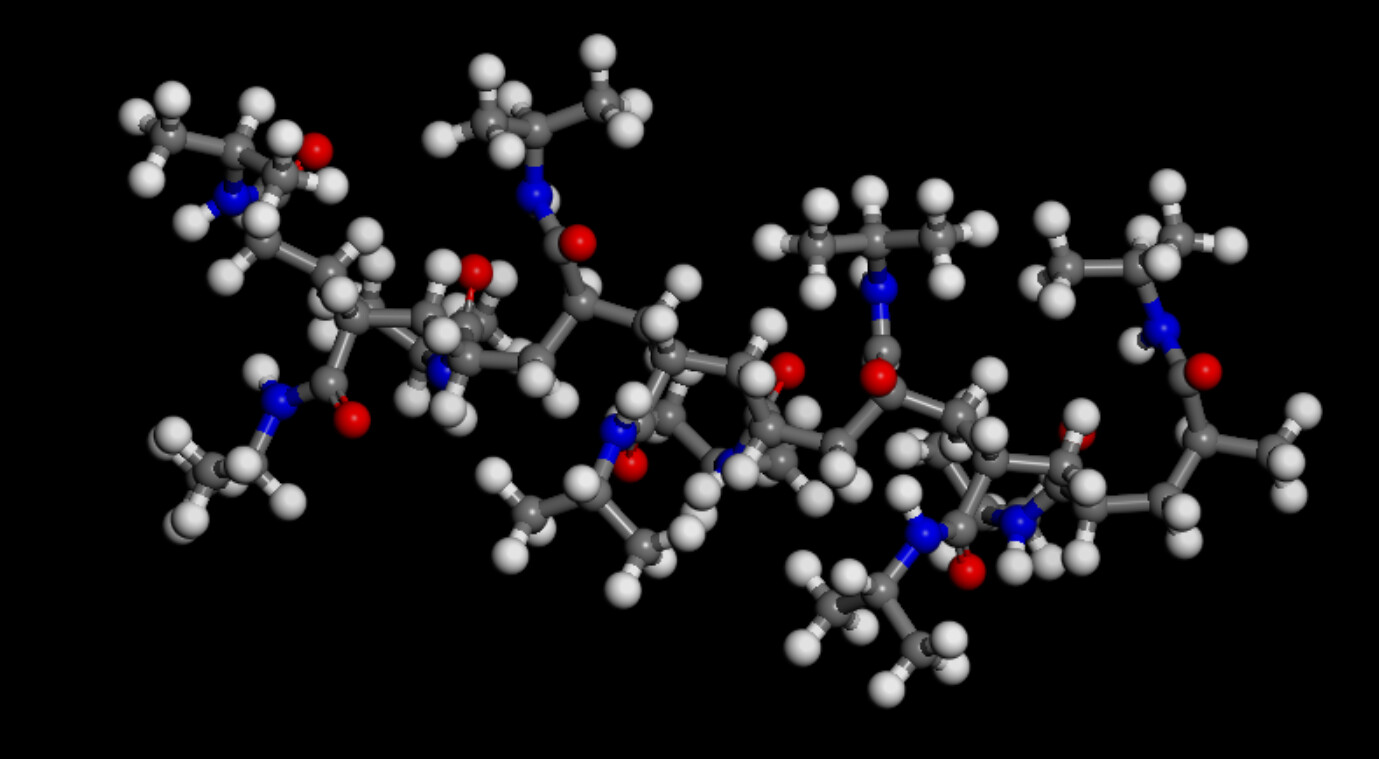
The problem was defining the water, Ca2+, and Cl- with their own atom types: @atom:O, @atom:H, @atom:Ca, and @atom:Cl on top of the atom types already defined in the oplsaa.lt file. This resulted in a mismatch between pair coefficients that produces lost atoms.
I was able to reproduce your error with this definition of the system (LT file):
import "H2O.lt"
import "ions.lt"
import "oplsaa.lt"
import "PolyNIPAM.lt"
# Create the system.
wat=new H2O[500]
pol=new PolyNIPAM[1]
cat=new Ca[1]
ani=new Cl[2]
With the imports above, the force field settings are specified more than once, as they are inherited by both H2O.lt and oplsaa.lt. The resulting file then must be edited manually after being created by Moltemplate.
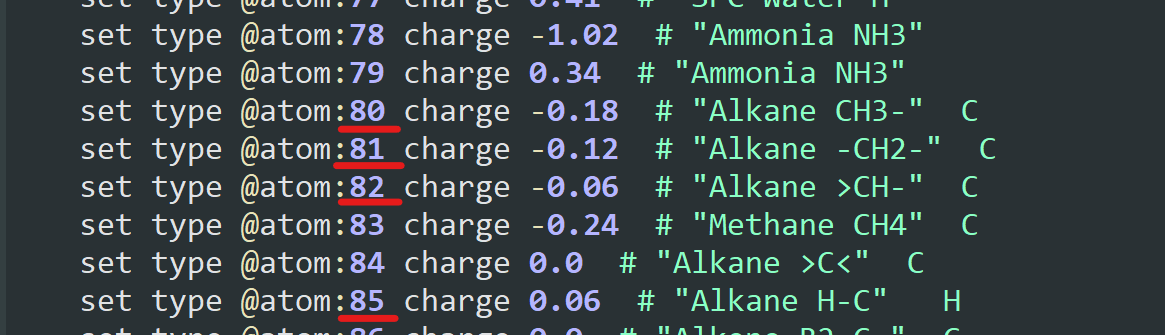
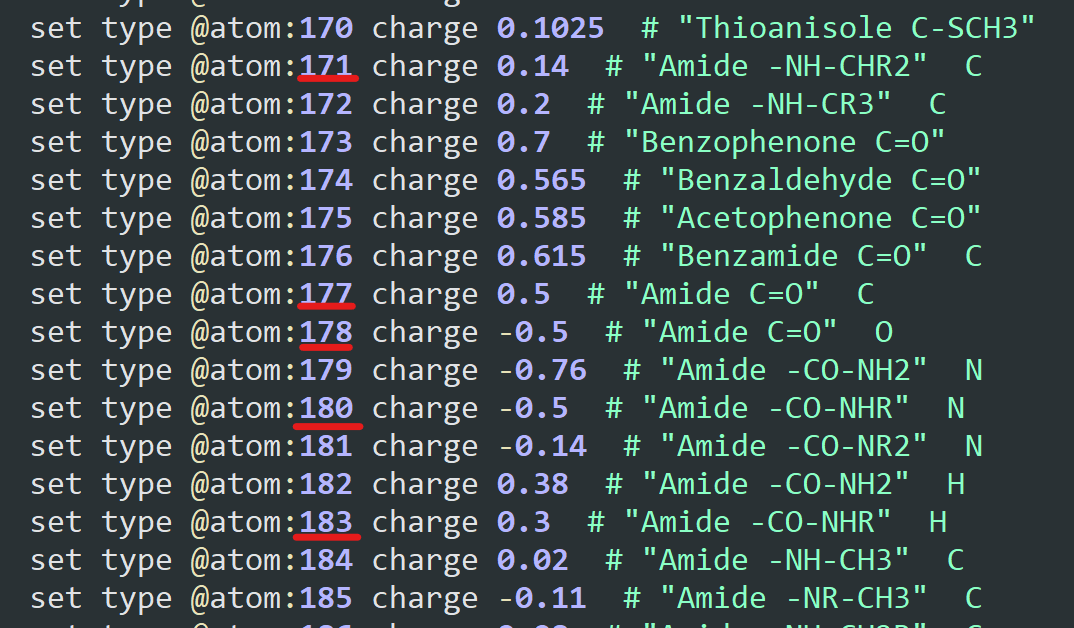
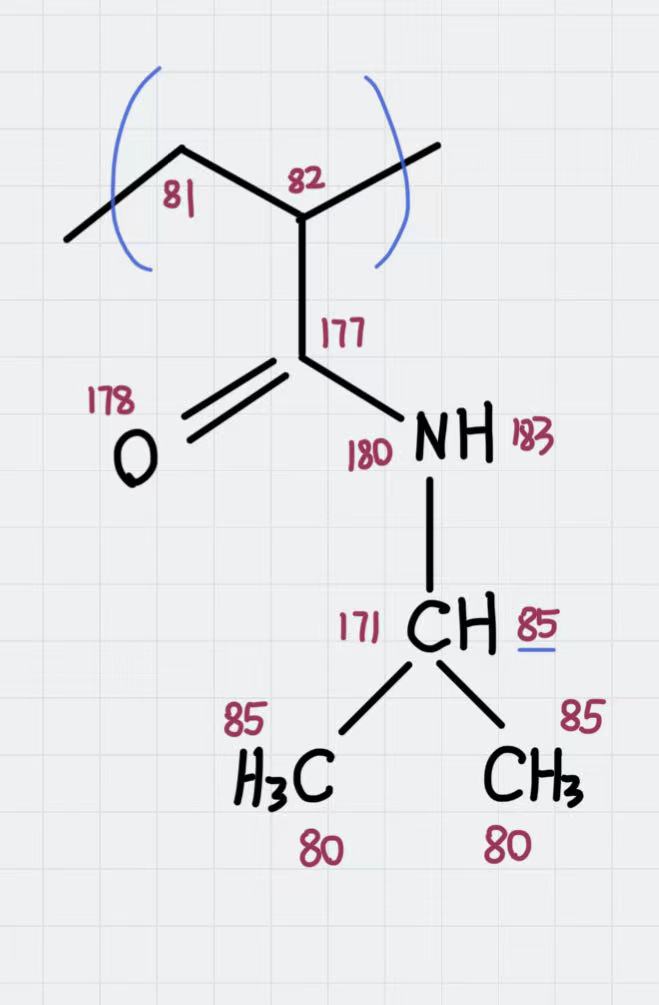
You were right: the atom types were assigned correctly for the polymer. Their value was just shifted as 4 new atom types were created before the set defined in OPLS-AA.
If instead of mixing different force fields you describe all the molecular species with OPLS-AA, then the simulation goes just fine. Here are my changes.
First of all, I have assigned a single molid to the polymer residue:
PolyNIPAM.lt
PolyNIPAM inherits OPLSAA {
# Charges will be overwritten.
write("Data Atoms") {
$atom:81_1 $mol @atom:81 0.0 -2.357 -0.245 0.629
$atom:81_2 $mol @atom:81 0.0 -1.194 0.752 0.778
$atom:85_1 $mol @atom:85 0.0 -2.027 -1.242 0.957
$atom:177_1 $mol @atom:177 0.0 -1.743 2.168 0.640
$atom:85_2 $mol @atom:85 0.0 -0.366 0.490 0.103
$atom:180_1 $mol @atom:180 0.0 -1.147 2.943 -0.274
$atom:178_1 $mol @atom:178 0.0 -2.700 2.545 1.293
$atom:171_1 $mol @atom:171 0.0 -1.602 4.322 -0.446
$atom:183_1 $mol @atom:183 0.0 -0.241 2.724 -0.647
$atom:80_1 $mol @atom:80 0.0 -2.662 4.430 -1.547
$atom:80_2 $mol @atom:80 0.0 -0.400 5.227 -0.741
$atom:85_3 $mol @atom:85 0.0 -2.053 4.672 0.501
$atom:85_4 $mol @atom:85 0.0 -3.539 3.796 -1.342
$atom:85_5 $mol @atom:85 0.0 -2.245 4.148 -2.526
$atom:85_6 $mol @atom:85 0.0 -3.006 5.475 -1.612
$atom:85_7 $mol @atom:85 0.0 0.080 4.944 -1.690
$atom:85_8 $mol @atom:85 0.0 0.355 5.185 0.060
$atom:85_9 $mol @atom:85 0.0 -0.737 6.273 -0.826
$atom:85_10 $mol @atom:85 0.0 -3.156 0.050 1.336
$atom:85_11 $mol @atom:85 0.0 -0.808 0.718 1.811
$atom:81_3 $mol @atom:81 0.0 -4.482 -0.662 -0.732
$atom:82_1 $mol @atom:82 0.0 -2.984 -0.308 -0.783
$atom:85_12 $mol @atom:85 0.0 -4.918 0.018 0.014
$atom:177_2 $mol @atom:177 0.0 -2.344 -1.080 -1.873
$atom:180_2 $mol @atom:180 0.0 -1.131 -1.604 -1.620
$atom:178_2 $mol @atom:178 0.0 -2.876 -1.219 -2.965
$atom:171_2 $mol @atom:171 0.0 -0.391 -2.304 -2.661
$atom:183_2 $mol @atom:183 0.0 -0.690 -1.494 -0.727
$atom:80_3 $mol @atom:80 0.0 0.802 -3.029 -2.033
$atom:80_4 $mol @atom:80 0.0 0.094 -1.373 -3.777
$atom:85_13 $mol @atom:85 0.0 -1.054 -3.067 -3.111
$atom:85_14 $mol @atom:85 0.0 0.481 -3.774 -1.288
$atom:85_15 $mol @atom:85 0.0 1.485 -2.319 -1.542
$atom:85_16 $mol @atom:85 0.0 1.368 -3.558 -2.817
$atom:85_17 $mol @atom:85 0.0 0.761 -0.594 -3.382
$atom:85_18 $mol @atom:85 0.0 -0.738 -0.879 -4.300
$atom:85_19 $mol @atom:85 0.0 0.653 -1.964 -4.521
$atom:85_20 $mol @atom:85 0.0 -4.937 -0.363 -1.697
$atom:85_21 $mol @atom:85 0.0 -2.967 0.717 -1.203
$atom:81_4 $mol @atom:81 0.0 -6.422 -2.235 -0.754
$atom:82_2 $mol @atom:82 0.0 -4.928 -2.114 -0.469
$atom:85_22 $mol @atom:85 0.0 -6.565 -1.832 -1.770
$atom:177_3 $mol @atom:177 0.0 -4.663 -2.569 0.992
$atom:180_3 $mol @atom:180 0.0 -3.960 -3.716 1.081
$atom:178_3 $mol @atom:178 0.0 -4.967 -1.904 1.963
$atom:171_3 $mol @atom:171 0.0 -3.354 -4.204 2.319
$atom:183_3 $mol @atom:183 0.0 -4.127 -4.397 0.356
$atom:80_5 $mol @atom:80 0.0 -4.295 -4.572 3.473
$atom:80_6 $mol @atom:80 0.0 -2.271 -3.244 2.828
$atom:85_23 $mol @atom:85 0.0 -2.826 -5.135 2.038
$atom:85_24 $mol @atom:85 0.0 -4.973 -5.396 3.211
$atom:85_25 $mol @atom:85 0.0 -4.909 -3.718 3.794
$atom:85_26 $mol @atom:85 0.0 -3.683 -4.896 4.331
$atom:85_27 $mol @atom:85 0.0 -2.695 -2.272 3.123
$atom:85_28 $mol @atom:85 0.0 -1.484 -3.073 2.079
$atom:85_29 $mol @atom:85 0.0 -1.791 -3.686 3.717
$atom:85_30 $mol @atom:85 0.0 -6.661 -3.312 -0.799
$atom:85_31 $mol @atom:85 0.0 -4.422 -2.764 -1.206
$atom:81_5 $mol @atom:81 0.0 -8.861 -2.101 -0.226
$atom:82_3 $mol @atom:82 0.0 -7.457 -1.604 0.194
$atom:85_32 $mol @atom:85 0.0 -8.826 -3.186 -0.048
$atom:177_4 $mol @atom:177 0.0 -7.425 -0.036 0.256
$atom:180_4 $mol @atom:180 0.0 -6.870 0.431 1.405
$atom:178_4 $mol @atom:178 0.0 -8.004 0.653 -0.558
$atom:171_4 $mol @atom:171 0.0 -7.168 1.765 1.912
$atom:183_4 $mol @atom:183 0.0 -6.797 -0.317 2.080
$atom:80_7 $mol @atom:80 0.0 -8.664 2.017 2.147
$atom:80_8 $mol @atom:80 0.0 -6.545 2.857 1.036
$atom:85_33 $mol @atom:85 0.0 -6.669 1.831 2.899
$atom:85_34 $mol @atom:85 0.0 -9.114 1.252 2.800
$atom:85_35 $mol @atom:85 0.0 -9.236 2.024 1.205
$atom:85_36 $mol @atom:85 0.0 -8.786 3.001 2.625
$atom:85_37 $mol @atom:85 0.0 -7.002 2.900 0.037
$atom:85_38 $mol @atom:85 0.0 -5.460 2.706 0.913
$atom:85_39 $mol @atom:85 0.0 -6.693 3.836 1.521
$atom:85_40 $mol @atom:85 0.0 -9.602 -1.708 0.496
$atom:85_41 $mol @atom:85 0.0 -7.328 -2.030 1.207
$atom:81_6 $mol @atom:81 0.0 -10.877 -2.392 -1.741
$atom:82_4 $mol @atom:82 0.0 -9.440 -1.848 -1.636
$atom:85_42 $mol @atom:85 0.0 -11.441 -2.051 -0.857
$atom:177_5 $mol @atom:177 0.0 -8.631 -2.553 -2.770
$atom:180_5 $mol @atom:180 0.0 -8.499 -1.785 -3.863
$atom:178_5 $mol @atom:178 0.0 -8.181 -3.684 -2.688
$atom:171_5 $mol @atom:171 0.0 -7.938 -2.258 -5.119
$atom:183_5 $mol @atom:183 0.0 -9.049 -0.943 -3.902
$atom:80_9 $mol @atom:80 0.0 -8.792 -3.342 -5.780
$atom:80_10 $mol @atom:80 0.0 -6.470 -2.687 -5.003
$atom:85_43 $mol @atom:85 0.0 -7.963 -1.383 -5.795
$atom:85_44 $mol @atom:85 0.0 -9.825 -2.995 -5.943
$atom:85_45 $mol @atom:85 0.0 -8.838 -4.264 -5.179
$atom:85_46 $mol @atom:85 0.0 -8.360 -3.594 -6.761
$atom:85_47 $mol @atom:85 0.0 -6.361 -3.605 -4.407
$atom:85_48 $mol @atom:85 0.0 -5.850 -1.903 -4.541
$atom:85_49 $mol @atom:85 0.0 -6.071 -2.887 -6.011
$atom:85_50 $mol @atom:85 0.0 -11.356 -1.921 -2.619
$atom:85_51 $mol @atom:85 0.0 -9.494 -0.756 -1.795
$atom:81_7 $mol @atom:81 0.0 -12.476 -4.326 -2.296
$atom:82_5 $mol @atom:82 0.0 -11.042 -3.918 -1.914
$atom:85_52 $mol @atom:85 0.0 -12.606 -4.028 -3.347
$atom:177_6 $mol @atom:177 0.0 -10.631 -4.675 -0.614
$atom:180_6 $mol @atom:180 0.0 -9.796 -5.703 -0.861
$atom:178_6 $mol @atom:178 0.0 -11.127 -4.453 0.474
$atom:171_6 $mol @atom:171 0.0 -9.186 -6.529 0.183
$atom:183_6 $mol @atom:183 0.0 -9.206 -5.554 -1.669
$atom:80_11 $mol @atom:80 0.0 -10.225 -7.315 0.995
$atom:80_12 $mol @atom:80 0.0 -8.204 -5.783 1.100
$atom:85_53 $mol @atom:85 0.0 -8.602 -7.290 -0.368
$atom:85_54 $mol @atom:85 0.0 -10.889 -7.898 0.339
$atom:85_55 $mol @atom:85 0.0 -10.843 -6.658 1.625
$atom:85_56 $mol @atom:85 0.0 -9.699 -8.020 1.659
$atom:85_57 $mol @atom:85 0.0 -8.739 -5.060 1.735
$atom:85_58 $mol @atom:85 0.0 -7.431 -5.245 0.530
$atom:85_59 $mol @atom:85 0.0 -7.700 -6.510 1.757
$atom:85_60 $mol @atom:85 0.0 -12.525 -5.430 -2.285
$atom:85_61 $mol @atom:85 0.0 -10.438 -4.244 -2.777
$atom:81_8 $mol @atom:81 0.0 -14.920 -4.584 -1.600
$atom:82_6 $mol @atom:82 0.0 -13.632 -3.752 -1.465
$atom:85_62 $mol @atom:85 0.0 -14.649 -5.586 -1.247
$atom:177_7 $mol @atom:177 0.0 -13.893 -2.285 -1.914
$atom:180_7 $mol @atom:180 0.0 -13.965 -1.422 -0.889
$atom:178_7 $mol @atom:178 0.0 -13.974 -1.956 -3.082
$atom:171_7 $mol @atom:171 0.0 -14.239 0.001 -1.025
$atom:183_7 $mol @atom:183 0.0 -13.577 -1.708 -0.004
$atom:80_13 $mol @atom:80 0.0 -15.680 0.246 -1.480
$atom:80_14 $mol @atom:80 0.0 -13.267 0.807 -1.893
$atom:85_63 $mol @atom:85 0.0 -14.156 0.414 -0.002
$atom:85_64 $mol @atom:85 0.0 -16.408 -0.251 -0.821
$atom:85_65 $mol @atom:85 0.0 -15.849 -0.114 -2.507
$atom:85_66 $mol @atom:85 0.0 -15.890 1.327 -1.461
$atom:85_67 $mol @atom:85 0.0 -13.268 0.469 -2.940
$atom:85_68 $mol @atom:85 0.0 -12.239 0.760 -1.513
$atom:85_69 $mol @atom:85 0.0 -13.597 1.859 -1.881
$atom:85_70 $mol @atom:85 0.0 -15.637 -4.195 -0.859
$atom:85_71 $mol @atom:85 0.0 -13.362 -3.817 -0.395
$atom:81_9 $mol @atom:81 0.0 -17.064 -5.395 -2.685
$atom:82_7 $mol @atom:82 0.0 -15.684 -4.733 -2.934
$atom:85_72 $mol @atom:85 0.0 -17.540 -4.759 -1.921
$atom:177_8 $mol @atom:177 0.0 -14.870 -5.540 -3.996
$atom:180_8 $mol @atom:180 0.0 -14.373 -4.756 -4.977
$atom:178_8 $mol @atom:178 0.0 -14.748 -6.750 -3.981
$atom:171_8 $mol @atom:171 0.0 -13.393 -5.268 -5.940
$atom:183_8 $mol @atom:183 0.0 -14.174 -3.807 -4.679
$atom:80_15 $mol @atom:80 0.0 -14.106 -6.091 -7.020
$atom:80_16 $mol @atom:80 0.0 -12.175 -6.042 -5.407
$atom:85_73 $mol @atom:85 0.0 -12.988 -4.367 -6.439
$atom:85_74 $mol @atom:85 0.0 -14.926 -5.528 -7.494
$atom:85_75 $mol @atom:85 0.0 -14.530 -7.019 -6.606
$atom:85_76 $mol @atom:85 0.0 -13.392 -6.372 -7.812
$atom:85_77 $mol @atom:85 0.0 -12.467 -7.007 -4.964
$atom:85_78 $mol @atom:85 0.0 -11.600 -5.477 -4.663
$atom:85_79 $mol @atom:85 0.0 -11.507 -6.251 -6.259
$atom:85_80 $mol @atom:85 0.0 -17.675 -5.258 -3.598
$atom:85_81 $mol @atom:85 0.0 -15.938 -3.724 -3.313
$atom:81_10 $mol @atom:81 0.0 -18.749 -7.194 -2.096
$atom:82_8 $mol @atom:82 0.0 -17.248 -6.883 -2.287
$atom:85_82 $mol @atom:85 0.0 -19.267 -6.796 -2.989
$atom:177_9 $mol @atom:177 0.0 -16.454 -7.269 -0.999
$atom:180_9 $mol @atom:180 0.0 -15.335 -7.967 -1.254
$atom:178_9 $mol @atom:178 0.0 -16.762 -6.899 0.120
$atom:171_9 $mol @atom:171 0.0 -14.367 -8.387 -0.248
$atom:183_9 $mol @atom:183 0.0 -15.240 -8.331 -2.192
$atom:80_17 $mol @atom:80 0.0 -14.904 -9.451 0.715
$atom:80_18 $mol @atom:80 0.0 -13.725 -7.242 0.549
$atom:85_83 $mol @atom:85 0.0 -13.545 -8.861 -0.817
$atom:85_84 $mol @atom:85 0.0 -15.290 -10.333 0.182
$atom:85_85 $mol @atom:85 0.0 -15.717 -9.064 1.350
$atom:85_86 $mol @atom:85 0.0 -14.087 -9.788 1.373
$atom:85_87 $mol @atom:85 0.0 -14.468 -6.647 1.100
$atom:85_88 $mol @atom:85 0.0 -13.139 -6.565 -0.089
$atom:85_89 $mol @atom:85 0.0 -13.036 -7.679 1.290
$atom:85_90 $mol @atom:85 0.0 -18.877 -8.292 -2.150
$atom:85_91 $mol @atom:85 0.0 -16.939 -7.505 -3.147
$atom:80_19 $mol @atom:80 0.0 -20.999 -7.206 -1.036
$atom:82_9 $mol @atom:82 0.0 -19.540 -6.757 -0.843
$atom:85_92 $mol @atom:85 0.0 -21.046 -8.300 -1.156
$atom:85_93 $mol @atom:85 0.0 -21.439 -6.747 -1.936
$atom:177_10 $mol @atom:177 0.0 -19.578 -5.207 -0.628
$atom:180_10 $mol @atom:180 0.0 -19.061 -4.828 0.557
$atom:178_10 $mol @atom:178 0.0 -20.185 -4.465 -1.375
$atom:171_10 $mol @atom:171 0.0 -18.878 -3.426 0.994
$atom:183_10 $mol @atom:183 0.0 -18.342 -5.450 0.901
$atom:80_20 $mol @atom:80 0.0 -19.984 -2.451 0.558
$atom:80_21 $mol @atom:80 0.0 -17.509 -2.857 0.610
$atom:85_94 $mol @atom:85 0.0 -18.906 -3.470 2.099
$atom:85_95 $mol @atom:85 0.0 -20.991 -2.887 0.651
$atom:85_96 $mol @atom:85 0.0 -19.829 -2.132 -0.485
$atom:85_97 $mol @atom:85 0.0 -19.955 -1.550 1.193
$atom:85_98 $mol @atom:85 0.0 -17.405 -2.773 -0.482
$atom:85_99 $mol @atom:85 0.0 -16.684 -3.475 0.993
$atom:85_100 $mol @atom:85 0.0 -17.407 -1.849 1.043
$atom:85_101 $mol @atom:85 0.0 -21.621 -6.939 -0.168
$atom:85_102 $mol @atom:85 0.0 -19.160 -7.294 0.044
}
write("Data Bond List") {
$bond:id1 $atom:81_1 $atom:81_2
$bond:id2 $atom:81_1 $atom:85_1
$bond:id3 $atom:81_1 $atom:85_10
$bond:id4 $atom:81_1 $atom:82_1
$bond:id5 $atom:81_2 $atom:177_1
$bond:id6 $atom:81_2 $atom:85_2
$bond:id7 $atom:81_2 $atom:85_11
$bond:id8 $atom:177_1 $atom:180_1
$bond:id9 $atom:177_1 $atom:178_1
$bond:id10 $atom:180_1 $atom:171_1
$bond:id11 $atom:180_1 $atom:183_1
$bond:id12 $atom:171_1 $atom:80_1
$bond:id13 $atom:171_1 $atom:80_2
$bond:id14 $atom:85_3 $atom:171_1
$bond:id15 $atom:85_4 $atom:80_1
$bond:id16 $atom:85_5 $atom:80_1
$bond:id17 $atom:85_6 $atom:80_1
$bond:id18 $atom:85_7 $atom:80_2
$bond:id19 $atom:85_8 $atom:80_2
$bond:id20 $atom:85_9 $atom:80_2
$bond:id21 $atom:81_3 $atom:82_1
$bond:id22 $atom:81_3 $atom:85_12
$bond:id23 $atom:81_3 $atom:85_20
$bond:id24 $atom:81_3 $atom:82_2
$bond:id25 $atom:177_2 $atom:82_1
$bond:id26 $atom:85_21 $atom:82_1
$bond:id27 $atom:177_2 $atom:180_2
$bond:id28 $atom:177_2 $atom:178_2
$bond:id29 $atom:180_2 $atom:171_2
$bond:id30 $atom:180_2 $atom:183_2
$bond:id31 $atom:171_2 $atom:80_3
$bond:id32 $atom:171_2 $atom:80_4
$bond:id33 $atom:85_13 $atom:171_2
$bond:id34 $atom:85_14 $atom:80_3
$bond:id35 $atom:85_15 $atom:80_3
$bond:id36 $atom:85_16 $atom:80_3
$bond:id37 $atom:85_17 $atom:80_4
$bond:id38 $atom:85_18 $atom:80_4
$bond:id39 $atom:85_19 $atom:80_4
$bond:id40 $atom:81_4 $atom:82_2
$bond:id41 $atom:81_4 $atom:85_22
$bond:id42 $atom:81_4 $atom:85_30
$bond:id43 $atom:81_4 $atom:82_3
$bond:id44 $atom:177_3 $atom:82_2
$bond:id45 $atom:85_31 $atom:82_2
$bond:id46 $atom:177_3 $atom:180_3
$bond:id47 $atom:177_3 $atom:178_3
$bond:id48 $atom:180_3 $atom:171_3
$bond:id49 $atom:180_3 $atom:183_3
$bond:id50 $atom:171_3 $atom:80_5
$bond:id51 $atom:171_3 $atom:80_6
$bond:id52 $atom:85_23 $atom:171_3
$bond:id53 $atom:85_24 $atom:80_5
$bond:id54 $atom:85_25 $atom:80_5
$bond:id55 $atom:85_26 $atom:80_5
$bond:id56 $atom:85_27 $atom:80_6
$bond:id57 $atom:85_28 $atom:80_6
$bond:id58 $atom:85_29 $atom:80_6
$bond:id59 $atom:81_5 $atom:82_3
$bond:id60 $atom:81_5 $atom:85_32
$bond:id61 $atom:81_5 $atom:85_40
$bond:id62 $atom:81_5 $atom:82_4
$bond:id63 $atom:177_4 $atom:82_3
$bond:id64 $atom:85_41 $atom:82_3
$bond:id65 $atom:177_4 $atom:180_4
$bond:id66 $atom:177_4 $atom:178_4
$bond:id67 $atom:180_4 $atom:171_4
$bond:id68 $atom:180_4 $atom:183_4
$bond:id69 $atom:171_4 $atom:80_7
$bond:id70 $atom:171_4 $atom:80_8
$bond:id71 $atom:85_33 $atom:171_4
$bond:id72 $atom:85_34 $atom:80_7
$bond:id73 $atom:85_35 $atom:80_7
$bond:id74 $atom:85_36 $atom:80_7
$bond:id75 $atom:85_37 $atom:80_8
$bond:id76 $atom:85_38 $atom:80_8
$bond:id77 $atom:85_39 $atom:80_8
$bond:id78 $atom:81_6 $atom:82_4
$bond:id79 $atom:81_6 $atom:85_42
$bond:id80 $atom:81_6 $atom:85_50
$bond:id81 $atom:81_6 $atom:82_5
$bond:id82 $atom:177_5 $atom:82_4
$bond:id83 $atom:85_51 $atom:82_4
$bond:id84 $atom:177_5 $atom:180_5
$bond:id85 $atom:177_5 $atom:178_5
$bond:id86 $atom:180_5 $atom:171_5
$bond:id87 $atom:180_5 $atom:183_5
$bond:id88 $atom:171_5 $atom:80_9
$bond:id89 $atom:171_5 $atom:80_10
$bond:id90 $atom:85_43 $atom:171_5
$bond:id91 $atom:85_44 $atom:80_9
$bond:id92 $atom:85_45 $atom:80_9
$bond:id93 $atom:85_46 $atom:80_9
$bond:id94 $atom:85_47 $atom:80_10
$bond:id95 $atom:85_48 $atom:80_10
$bond:id96 $atom:85_49 $atom:80_10
$bond:id97 $atom:81_7 $atom:82_5
$bond:id98 $atom:81_7 $atom:85_52
$bond:id99 $atom:81_7 $atom:85_60
$bond:id100 $atom:81_7 $atom:82_6
$bond:id101 $atom:177_6 $atom:82_5
$bond:id102 $atom:85_61 $atom:82_5
$bond:id103 $atom:177_6 $atom:180_6
$bond:id104 $atom:177_6 $atom:178_6
$bond:id105 $atom:180_6 $atom:171_6
$bond:id106 $atom:180_6 $atom:183_6
$bond:id107 $atom:171_6 $atom:80_11
$bond:id108 $atom:171_6 $atom:80_12
$bond:id109 $atom:85_53 $atom:171_6
$bond:id110 $atom:85_54 $atom:80_11
$bond:id111 $atom:85_55 $atom:80_11
$bond:id112 $atom:85_56 $atom:80_11
$bond:id113 $atom:85_57 $atom:80_12
$bond:id114 $atom:85_58 $atom:80_12
$bond:id115 $atom:85_59 $atom:80_12
$bond:id116 $atom:81_8 $atom:82_6
$bond:id117 $atom:81_8 $atom:85_62
$bond:id118 $atom:81_8 $atom:85_70
$bond:id119 $atom:81_8 $atom:82_7
$bond:id120 $atom:177_7 $atom:82_6
$bond:id121 $atom:85_71 $atom:82_6
$bond:id122 $atom:177_7 $atom:180_7
$bond:id123 $atom:177_7 $atom:178_7
$bond:id124 $atom:180_7 $atom:171_7
$bond:id125 $atom:180_7 $atom:183_7
$bond:id126 $atom:171_7 $atom:80_13
$bond:id127 $atom:171_7 $atom:80_14
$bond:id128 $atom:85_63 $atom:171_7
$bond:id129 $atom:85_64 $atom:80_13
$bond:id130 $atom:85_65 $atom:80_13
$bond:id131 $atom:85_66 $atom:80_13
$bond:id132 $atom:85_67 $atom:80_14
$bond:id133 $atom:85_68 $atom:80_14
$bond:id134 $atom:85_69 $atom:80_14
$bond:id135 $atom:81_9 $atom:82_7
$bond:id136 $atom:81_9 $atom:85_72
$bond:id137 $atom:81_9 $atom:85_80
$bond:id138 $atom:81_9 $atom:82_8
$bond:id139 $atom:177_8 $atom:82_7
$bond:id140 $atom:85_81 $atom:82_7
$bond:id141 $atom:177_8 $atom:180_8
$bond:id142 $atom:177_8 $atom:178_8
$bond:id143 $atom:180_8 $atom:171_8
$bond:id144 $atom:180_8 $atom:183_8
$bond:id145 $atom:171_8 $atom:80_15
$bond:id146 $atom:171_8 $atom:80_16
$bond:id147 $atom:85_73 $atom:171_8
$bond:id148 $atom:85_74 $atom:80_15
$bond:id149 $atom:85_75 $atom:80_15
$bond:id150 $atom:85_76 $atom:80_15
$bond:id151 $atom:85_77 $atom:80_16
$bond:id152 $atom:85_78 $atom:80_16
$bond:id153 $atom:85_79 $atom:80_16
$bond:id154 $atom:81_10 $atom:82_8
$bond:id155 $atom:81_10 $atom:85_82
$bond:id156 $atom:81_10 $atom:85_90
$bond:id157 $atom:81_10 $atom:82_9
$bond:id158 $atom:177_9 $atom:82_8
$bond:id159 $atom:85_91 $atom:82_8
$bond:id160 $atom:177_9 $atom:180_9
$bond:id161 $atom:177_9 $atom:178_9
$bond:id162 $atom:180_9 $atom:171_9
$bond:id163 $atom:180_9 $atom:183_9
$bond:id164 $atom:171_9 $atom:80_17
$bond:id165 $atom:171_9 $atom:80_18
$bond:id166 $atom:85_83 $atom:171_9
$bond:id167 $atom:85_84 $atom:80_17
$bond:id168 $atom:85_85 $atom:80_17
$bond:id169 $atom:85_86 $atom:80_17
$bond:id170 $atom:85_87 $atom:80_18
$bond:id171 $atom:85_88 $atom:80_18
$bond:id172 $atom:85_89 $atom:80_18
$bond:id173 $atom:80_19 $atom:82_9
$bond:id174 $atom:85_92 $atom:80_19
$bond:id175 $atom:85_93 $atom:80_19
$bond:id176 $atom:85_101 $atom:80_19
$bond:id177 $atom:177_10 $atom:82_9
$bond:id178 $atom:85_102 $atom:82_9
$bond:id179 $atom:177_10 $atom:180_10
$bond:id180 $atom:177_10 $atom:178_10
$bond:id181 $atom:180_10 $atom:171_10
$bond:id182 $atom:180_10 $atom:183_10
$bond:id183 $atom:171_10 $atom:80_20
$bond:id184 $atom:171_10 $atom:80_21
$bond:id185 $atom:85_94 $atom:171_10
$bond:id186 $atom:85_95 $atom:80_20
$bond:id187 $atom:85_96 $atom:80_20
$bond:id188 $atom:85_97 $atom:80_20
$bond:id189 $atom:85_98 $atom:80_21
$bond:id190 $atom:85_99 $atom:80_21
$bond:id191 $atom:85_100 $atom:80_21
}
} # end of "PolyNIPAM inherits OPLSAA" type definition
No need to specify the charges, as they will be overwritten.
Then I created a master file for the simulation. This file contains also the macros defining SPC water, Ca and Cl ions, but of course these species could be saved in separate files and imported. I put everything together for the sake of space.
sample01.lt
# Use the OPLS-AA force field for all species.
import "oplsaa.lt"
import "PolyNIPAM.lt"
# Define the SPC water and ions as in the OPLS-AA
Ca inherits OPLSAA {
write("Data Atoms"){
$atom:a1 $mol:. @atom:354 0.0 0.00000 0.00000 0.000000
}
}
Cl inherits OPLSAA {
write("Data Atoms"){
$atom:a1 $mol:. @atom:344 0.0 0.00000 0.00000 0.000000
}
}
H2O inherits OPLSAA {
write("Data Atoms"){
$atom:O $mol:. @atom:76 0. 0.0000000 0.00000 0.000000
$atom:H1 $mol:. @atom:77 0. 0.8164904 0.00000 0.5773590
$atom:H2 $mol:. @atom:77 0. -0.8164904 0.00000 0.5773590
}
write("Data Bond List") {
$bond:OH1 $atom:O $atom:H1
$bond:OH2 $atom:O $atom:H2
}
}
# Create the system.
wat=new H2O[500]
pol=new PolyNIPAM[1]
cat=new Ca[1]
ani=new Cl[2]
# Periodic boundary conditions:
write_once("Data Boundary"){
0 26 xlo xhi
0 26 ylo yhi
0 26 zlo zhi
}
# Define the input variables.
write_once("In Init"){
# Input variables.
variable ts equal 1 # timestep
variable temp equal 298.15 # equilibrium temperature
variable p equal 1. # equilibrium pressure
# PBC (set them before the creation of the box).
boundary p p p
neighbor 3 bin
}
# Let's go, Pickacku!
write_once("In Run"){
dump 33 all dcd 500 sample01.dcd
dump_modify 33 unwrap yes
group watergroup type 76 77
velocity all create \$\{temp\} 12345
timestep \$\{ts\}
thermo 1000
thermo_style custom step temp press pe ke etotal atoms lx ly lz vol density
thermo_modify flush yes
minimize .01 .001 1000 100000
write_data min.data
fix 0 watergroup shake 0.0001 20 0 b 1 a 1
fix 1 all nvt temp \$\{temp\} \$\{temp\} \$(100*dt) drag 2
run 500000
unfix 1
fix 1 all npt temp \$\{temp\} \$\{temp\} \$(100*dt) iso \$p \$p \$(1000*dt) drag 2
run 500000
unfix 1
write_data Equil.data
fix 1 all nvt temp \$\{temp\} \$\{temp\} \$(100*dt) drag 2
run 1000000
write_data final.data
}
Render with:
moltemplate.sh -overlay-all -pdb model.pdb sample02.lt
Run with:
mpirun -np 4 lmp_2Aug23 -in sample01.in -l sample01.log
In this case, the simulation quickly reaches thermal equilibrium:
I hope this is a helpful reference for anyone willing to use the OPLS-AA force field in combination with a PDB containing the initial guess of a molecular system.