Hi lammps users,
To clarify from previous messages, I realize that my issue is that I want to apply bonding terms on top of the Tersoff potential. This is not allowed, according to:
http://lammps.sandia.gov/doc/special_bonds.html
I realize that there is good reason to exclude many-body pair styles and bonding terms, as discussed in previous messages:
http://lammps.sandia.gov/threads/msg38173.html
http://lammps.sandia.gov/threads/msg38174.html
However, I believe this model (Tersoff + bonds) is physically reasonable, and necessary, for the stability of the PCBM molecule I am trying to study (also attached):
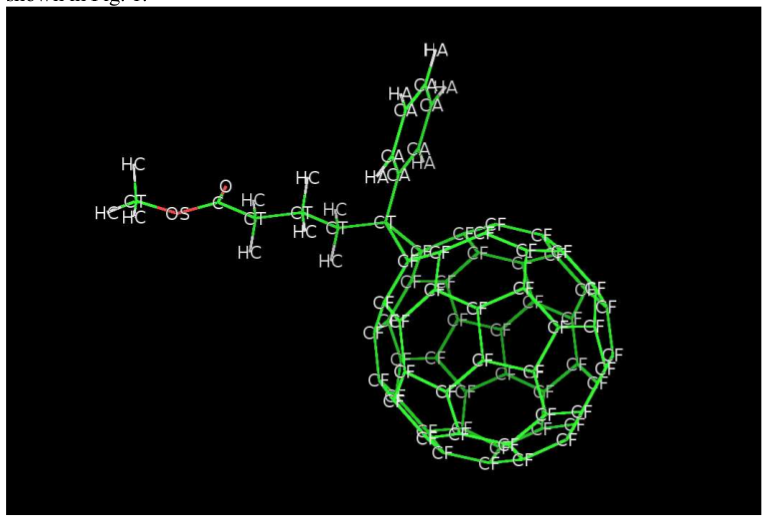
https://raw.github.com/jasonlarkin/disorder/master/pcbm/pcbm_pdb/vmd_output/pcbm_cheung.png
{kind=link}
The reason to use Tersoff is so I do not have to produce the (large) list of bonds for the 5- and 6-member rings of the C60 (atoms labeled CF in pcbm_cheung.png).
The reason to use bonds on top of the Tersoff potential is to secure the satellite molecule to the C60, where the required bonds are between, for example, CF-CT-CA, CF-CT-CT, CF-CF-CT-CA, etc.
I have tested this potential using the lattice/molecular dynamics program GULP, and the system seems to be stable.
Is it possible to implement Tersoff+bonds in some other way with the current version of LAMMPS?
If not, is there an easy way to modify the source code for my own purpose, and which routine(s) do I need to modify?
Thanks, and sorry for any previous confusion,
Jason