Hi all-
I’ve recently been trying to model some polymer systems using OPLS-AA in LAMMPS using EMC to build my initial structures. I had been trying to incorporate the new OPLS/2020 files which are reported in J. Phys. Chem. B 2024, 128, 1, 250–262, though that has proven to be a bit beyond my current skill level. (If anyone has had luck with this let me know)
My question is: when EMC spits out a .params file for LAMMPS from its build with OPLS, it chooses the harmonic, harmonic, harmonic, multi/harmonic, and harmonic styles for bonds, angles, dihedrals, and impropers, respectively. The bonds and angles I get, these match the form of OPLS. But for the dihedrals and impropers, the form specified in OPLS is a four-term cosine series,
for both types. The multi/harmonic dihedral potential in LAMMPS is given by
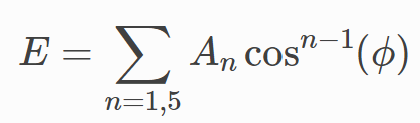
and the harmonic improper potential by
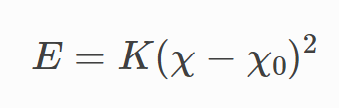
which are both different from the four-term cosine series. Is there a reason why EMC defaults to those potentials, and is it because they have been worked out to be mathematically equivalent to the original form? I feel like the harmonic improper one especially couldn’t really match the periodic cosine expression no matter what, unless there’s some math I’m forgetting.
In LAMMPS, it seems like it would make the most sense to use the dihedral_style/opls potential
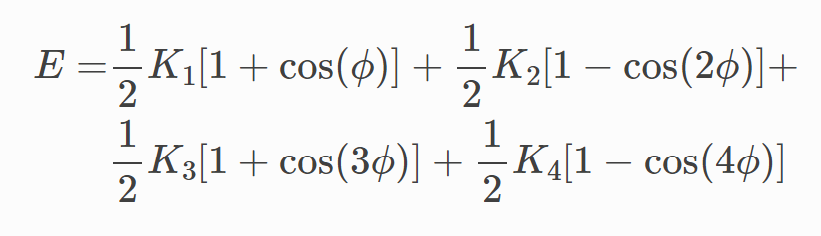
and the improper_style/cvff potential
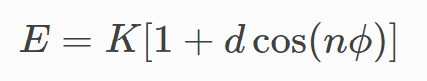
instead. Is there a way to get EMC to output these by default, or would this have to be changed manually?
Thank you so much,
Sam