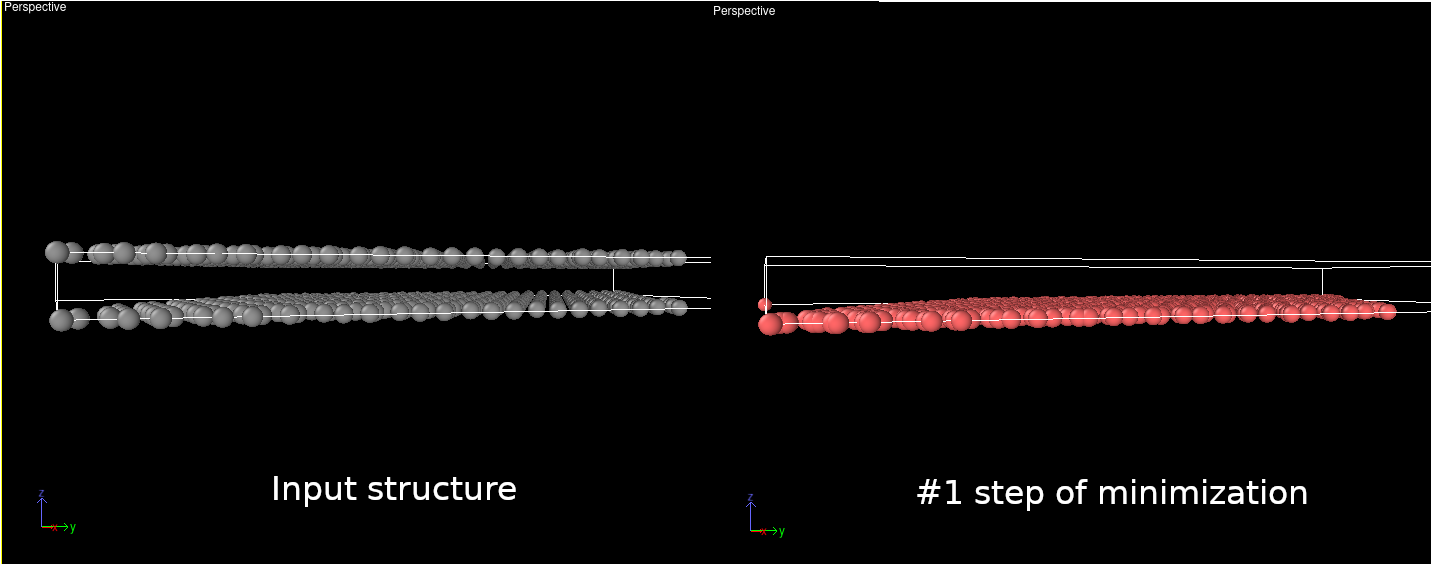
I’m a new user of LAMMPS. I believe I’ve used the correct input file to describe the 2 layer graphene structure. However, the result of energy minimization makes the graphene system shrink into 1 layer (as I see from the dump file). What is the best way to keep it 2 layers, or any command that I miss? I attached the files for you to take a look at.
I’ve tried changing the force field and limiting the interaction (setting r cut < interlayer distance, so the layers don’t shrink), but the result is still the same. Is there a way to set the group of atoms in a fixed position on the z-axis? Because I think this structure collapsed from the start, even before the minimization process
If you had a proper force field choice and proper parameters, the simulation will give you the expected structure. In fact, it is one of the crucial tests for force field parameterization that they can reproduce structures.
Neither is a plain lj/cut without bonds/angles/dihedrals is suitable to represent graphene, nor do your choice of cutoff and epsilon/sigma resemble typical parameters for covalent carbon atoms in metal units.
Thus the GI-GO rule applies: garbage in → garbage out.
What you are asking for makes not sense, because if you try “fix” a bad force field choice and parameters with disallowing the system where it “wants” to go, then you there is no meaning in doing a simulation in the first place.
Typical force field choices for graphene are either parameters for benzene carbon atoms from “bio” force fields like Amber, CHARMM, etc. which require a proper bond topology for directly connected atoms or manybody potentials like Tersoff or AIREBO or even something like ReaxFF with a suitable parameter set (ReaxFF parameters are very specific to the system they need to be used for. and setting up and running such calculations is the most difficult and time consuming option)