Dear All
I’m working on polyethylene crystallization, and now I want to analyze the transformation of crystal types during crystallization.
-
I used compute orientorder/atom command. My part in file is as follows:
-
-
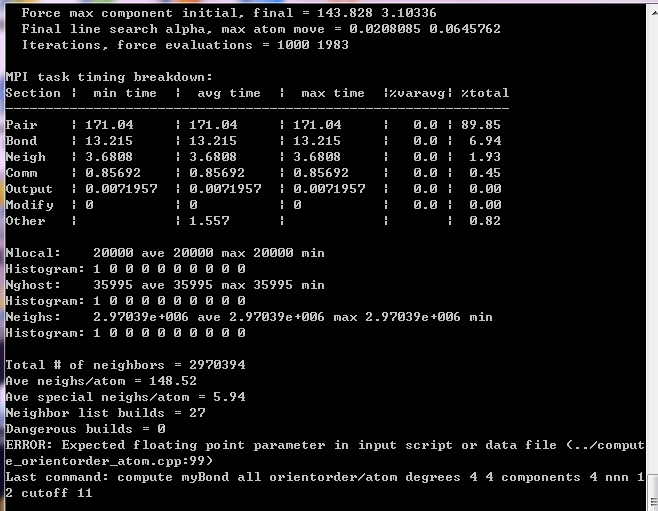
But I always report an error when I run it, and I didn’t find it in the manual.The error message is as follows:
I would really appreciate it if you could help me point out the mistakes and correct them
[
15584345983@…127…
|
](头像签名)
签名由 网易邮箱大师 定制
Dear All
I’m working on polyethylene crystallization, and now I want to analyze the transformation of crystal types during crystallization.
- But I always report an error when I run it, and I didn’t find it in the manual.The error message is as follows:
I would really appreciate it if you could help me point out the mistakes and correct them
please check the documentation more carefully. specifically look at the “degrees” keyword. your input states, that you want to compute 4 different order parameters, but you only provide one. so LAMMPS will try interpret the “components” word as a number, and stops with an error since it is not a number.
axel.

{kind=link}