This is a follow-up on a previous post but has a new facet that merits a post of its own. I am working with the same system described in the aforementioned post, but I’m running it on a GPU. Even with a timestep of 0.1 fs, I am still getting the “out of range atoms - cannot compute pppm” error. I have implemented both respa and shake.
Input files, output and error files, and the few trajectory files produced are available via the protondrive link below.
Any thoughts on how I can address this would be appreciated.
Not really. The origin of your problems is exactly the same.
That is redundant and thus pointless. Moreover, in this combination the pressure is not reliable and thus will lead to wrong results with fix npt.
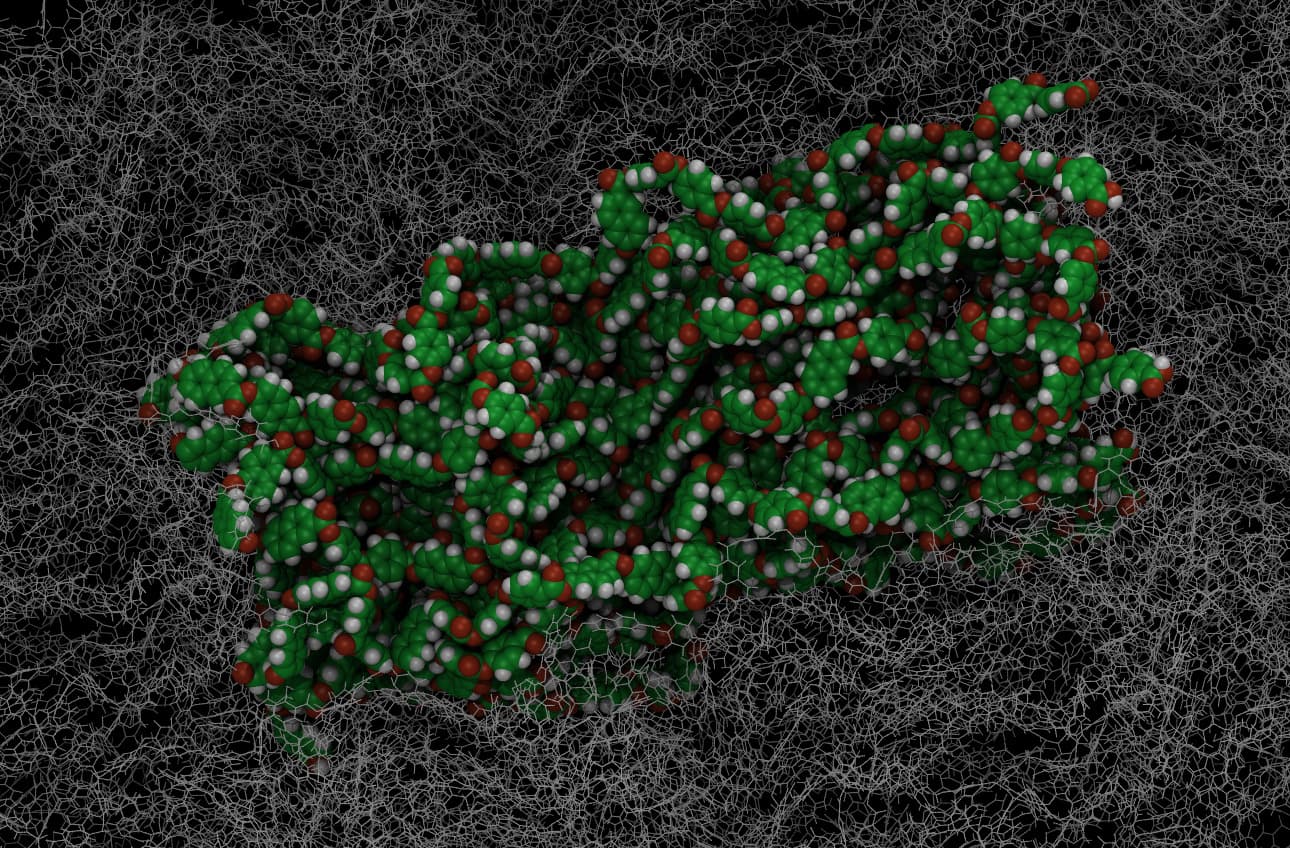
You have to address the issue that your initial geometry and box is very different from the equilibrated structure and thus has to undergo many transitions. Every time you have such a transition, a lot of potential energy is released (in layman’s terms, the system “snaps” into a new configuration) and that leads to large forces and thus atoms moving too fast and thus outside of range before the neighbor lists are updated. This process is made more difficult by using a far too large initial box and by using a very high temperature. I’ve manually edited the box dimensions, used regular verlet timestepping with shake and a timestep of 0.25 at 500 K target temperature. When alternating between minimizations and short MD runs (<= 1000 steps), it is possible to bring the system closer to equilibrium where it would be possible to run without the minimization quenches and then cool down to a more reasonable temperature and - finally - raise the timestep to 1.0fs or even 2.0fs. Also, when running on a single CPU with a single GPU, the simulation will be less likely to crash, because there are far fewer areas where atoms can get out of range. With this, I was able to run for quite a bit until I lost my patience.
To illustrate my point, I am attaching an initial geometry and the latest before I stopped.
After a considerable amount of simulation time, this system is still quite far from equilibrium, so out-of-range errors following some more transitions (or “snaps”) are still quite possible and the whole process will take an irritatingly long time. That is where Monte Carlo methods have their advantages.
My suggestion is thus to start over, and use a more effective way to build such a system. One such software, specifically designed for building polymer systems is called EMC. It also has its discussion forum here on MatSci.org.
VMD with a bit of post-processing and VMD scripting. Also, I changed the dump output from individual .xyz files to a single atom style LAMMPS native dump file.