Dear users,
I simulate water moelcules using:
velocity all create 1.0 12345689 dist uniform
fix 2 all npt temp 298.0 298.0 100.0 iso 1.0 1.0 100.0
timestep 0.5
run 100000
However, in this situation, the simulation will occur an error “Out of range atoms - cannot compute PPPM” at 42900 steps. While I change my input script into
velocity all create 298.0 12345689 dist uniform
fix 2 all npt temp 298.0 298.0 100.0 iso 1.0 1.0 100.0
timestep 0.5
run 100000
The simulation runs perfectly. Does any one have any idea about this? Thank you.
Miao-Chun
Dear users,
I simulate water moelcules using:
velocity all create 1.0 12345689 dist uniform
fix 2 all npt temp 298.0 298.0 100.0 iso 1.0 1.0 100.0
timestep 0.5
run 100000
However, in this situation, the simulation will occur an error "Out of
range atoms - cannot compute PPPM" at 42900 steps. While I change my input
script into
velocity all create 298.0 12345689 dist uniform
fix 2 all npt temp 298.0 298.0 100.0 iso 1.0 1.0 100.0
timestep 0.5
run 100000
The simulation runs perfectly. Does any one have any idea about this?
Thank you.
not from this little information. most likely you are doing something
wrong and it will eventually crash with both inputs. 100000 steps is not a
lot and with the different initialization, you will generate diverging
trajectories. furthermore, the first system will start from low temperature
and thus have to warm up, which takes time, especially since you have a
rather long thermostat time constant, so the probability in the first case
to have a situation happen that will cause the problem is less likely.
carefully visualize your trajectory and most likely you will see two atoms
getting very close and then shooting through the system at very high
velocity.
axel.
Dear users,
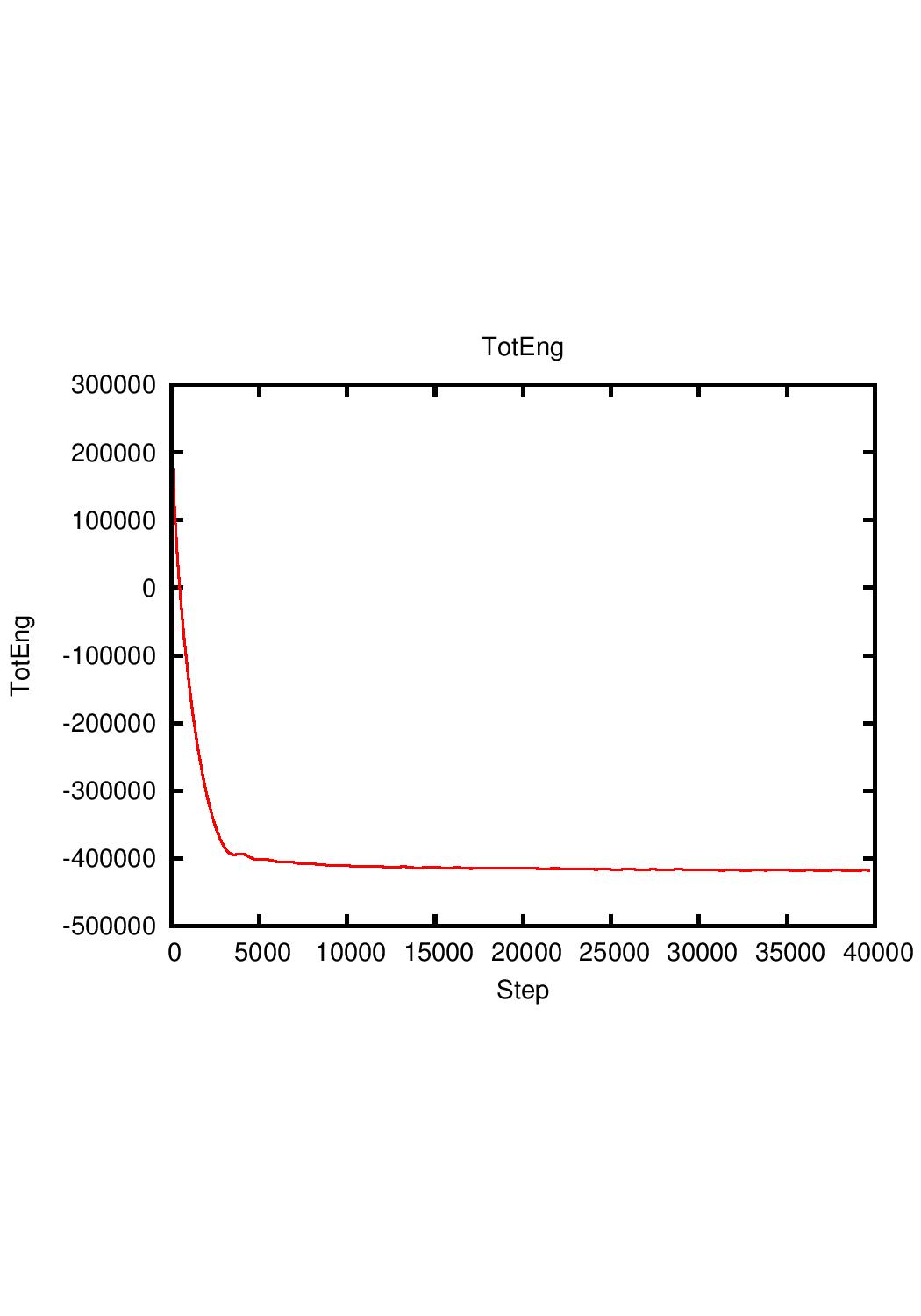
Above is the curve of total energy-steps of the water simulation for
velocity all create 298.0 12345689 dist uniform
fix 2 all npt temp 298.0 298.0 100.0 iso 1.0 1.0 100.0.
I agree that the first case happening the situation should be less probable than the second case.
I thought that looking over the total energy can prove that the simulation has been minimized, so by using 100000 steps I thought it is enough while the curve of total energy-steps at over 10000 steps has a slope approximately to zero. But apparently, if lammps cannot compute PPPM, then it means that the atom has gone too far to calculate, and this error result conflicts with what I thought. Do I make a mistake here?
However, I do not see atoms shooting in fast speed after visualize my simulation. But thank you Axel for pointing out another path for me, I will keep searching any atoms acting in high speed.
Below is my input script.
units real
dimension 3
boundary p p p
atom_style full
read_data system.data
group ox type 1
group hy type 2
set group ox charge -0.8476
set group hy charge 0.4238
Flexible SPC/E Potential Parameters
Zhang et al., Fluid Phase Equilibria, 262 (2007) 210-216
pair_style lj/cut/coul/long 10.0
pair_coeff 1 1 0.1502629 3.1169
pair_coeff 1 2 0.0341116368 2.04845
pair_coeff 2 2 0.00774378 0.98
bond_style harmonic
bond_coeff 1 176.864 0.9611
angle_style harmonic
angle_coeff 1 42.1845 109.4712
kspace_style pppm 1.0e-5
velocity all create 298.0 12345689 dist uniform
fix 2 all npt temp 298.0 298.0 100.0 iso 1.0 1.0 100.0
neighbor 2.0 bin
neigh_modify delay 0 every 10 check yes
thermo 100
thermo_style custom step temp press vol etotal
thermo_modify norm no flush yes
#dumps
dump waterdump all atom 100 298K.dump
#run variables
timestep 0.5
run 100000
Thank you.
Miao-Chun
neighbor 2.0 bin
neigh_modify delay 0 every 10 check yes
thermo 100
thermo_style custom step temp press vol etotal
thermo_modify norm no flush yes
#dumps
dump waterdump all atom 100 298K.dump
#run variables
timestep 0.5
you have a *very* aggressive time step for using a flexible water
potential without redistributing weights or simulating D2O instead of H2O.
try 0.25 fs
also, your neighbor list settings with "every 10", especially in
combination with "check yes", is aggressive, too. the gain of performance
going from rebuilding neighborlists after 10 steps over after 20 steps is
miniscule, but 20 steps may be too much and you could have a fast moving
atom sudden pop up.
with "check yes", it is much better to use "every 1" or "every 2", and
perhaps "delay 2" or "delay 4". this will be much safer and still offer
good performance.
axel.
Dear users,
Thank you Axel, I am running my simulation now, and so far it looks good.
About the time step, actually I do not know how to determine whether it is an aggressive time step or not. Is there any method?
Thank you.
Miao-Chun
Dear users,
Thank you Axel, I am running my simulation now, and so far it looks good.
About the time step, actually I do not know how to determine whether it is
an aggressive time step or not. Is there any method?
in the case of water, i just know this from 20+ years experience.
in general, you can estimate this from very simple principles: the faster
atoms move, the smaller the time step has to be. how fast atoms move
depends on atom masses, force constants, temperature and method of time
integration. since hydrogen atoms are the lightest, they tend to be moving
the fastest. if you replace them with deuterium, they'll be slower by a
factor of sqrt(2). if the force constant for the harmonic bond is doubled,
they'll be faster by a factor of sqrt(2). you can see this from the
relation of the reduced time unit to mass and energy in lj units.
you can typically see how aggressive you are from looking at energy
conservation, if you run an equilibrated system with fix nve without any
thermostat or barostat. with an agressive timestep, you get a significant
drift in the energy. ...and then you may still run into problems, if you
have some kind of meta-stable conformation that will run fine for a long
time and suddenly drop into a different potential trough and become very
fast due to the kinetic energy that is set free in the process. so good
choices, especially during equilibration, are usually very conservative.
0.25 fs is a good choice for most flexible water models. 0.5fs is
aggressive and works for some systems at low temperature. 1fs practically
never works unless you do some tricks. in case you use fix shake or fix
rattle, the fastest motions are removed and then 1fs is a conservative
choice, 2fs works in most cases and 2.5fs is aggressive. if you use a fix
rigid integrator, you are back to 0.1-0.25fs, since the time integration of
the discretized rotation is not so stable as the time integration for
translation. ...and so on and so forth.
axel.