I’m trying to store and output the z direction force acting a frozen water molecule (the water was only froze on z direction ), The frozen water molecule was specified as group ‘freezewater’. All of the water in my system was specified as group ‘water’. The id numbers of H and O in that water were 6718, 6719 and 6720. This is the main body in my input script:
By using this input script, I always got the error: ERROR: Out of range atoms - cannot compute PPPM.
Only if I turned off the pair interaction between group ‘water’ and group ‘freezewater ’ by activating the command line ’ neigh_modify exclude group freezewater water’, I can get rid of that error. However, I think the subsequently calculated force wasn’t the one that I really needed because of the excluded pair interaction.
How can I correctly calculated the z-direction force acting on the water molecule ?
If you use fix store/force you will store the
force on the group atoms before the setforce
command clears it out. You can then sum
it and output it, for example.
That's due to bad dynamics. Your atoms are moving
too far before you reneighbor and PPPM can't map
their charge to the FFT grid. Reneighbor more aggressively.
Instead of setforce command, I tried recenter command to fix the center of mass for the water molecule along z direction. I think this kind of method should be more close to the dynamic that I want.
The following is the main part of my input script.
fix 2 water nvt temp 300.0 300.0 100.0
compute COM freezewater com
fix onewater freezewater recenter NULL NULL 24.5 units box
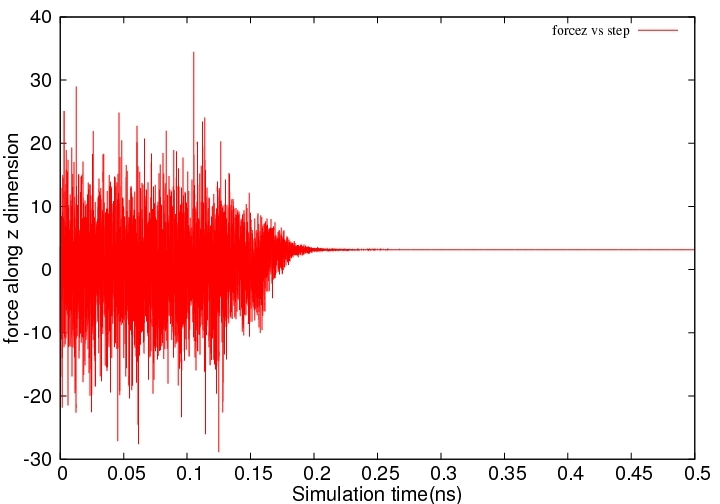
However, I got very strange result that I can’t understand. Plead find attachment. It is a figure for force applying on that water molecule along z dimension as a function of simulation time. Before 0.17 ns, everything looks good. But the force became about zero after about 0.17ns. Do you know why is that? I suppose the value of force should always fluctuate during dynamic simulation.
Instead of setforce command, I tried recenter command to fix the center of
mass for the water molecule along z direction. I think this kind of method
should be more close to the dynamic that I want.
The following is the main part of my input script.
fix 2 water nvt temp 300.0 300.0 100.0
compute COM freezewater com
fix onewater freezewater recenter NULL NULL 24.5 units box
Have you looked at (visualized) the trajectory using VMD? (or AtomEye
or some other program?) (If not, see attached README file containing
instructions for VMD.)
From your graph, it seems like the other water molecules are no longer
in contact with your frozen molecule. Did the other water molecules
move away from it?
In the manual, It is suggested that the reason for the decreasing of force is possibly due to the unpredictable dynamic of that freezing water if I didn’t use shift keyword to adjust the position of all atoms: ‘If you are thermostatting the entire system, then the solvent would be cooled to compensate’.
As you suggested, I checked the coordinate of that water but I didn’t find the other water move away. Instead, the water was froze on x,y,z dimensions after 0.17 ns.
I have tried not to integrate the equations of motion for the water that I want to fix. Just as you said, x,y,z coordinate of that water will be fixed during the simulation. But this is not what I want to do. I would like to only fix the z dimensional center of mass for that water and output the ‘real’ force on the water.
I found another command in the manual: ‘fix momentum’.
fix freeze freezewater momentum 1 linear 0 0 1
I hope the above command is able to zero out the linear momentum(velocity) of the water. Also, I hope the center of mass for that water won’t change with time. When I check the result of center of mass for that water, I found the center of mass along z dimension for the water changing with simulation time. But the range of changing was very small.
What I’m concerned is if this command will change the total force on that water. How did the ‘fix momentum’ command work? Can I get the true force on that water using this command?