Hello,
thanks for the message.
The problem is easy to fix and is not related to the actual implementation but the input.
In the input the kinetic scalar for the pressure calculation is incorrectly defined.
In the core/shell model, the pressure can be computed in a special manner by decoupling the
core/shell particles or in the normal LAMMPS convention
(J. Phys. Condens. Matter 6, 1994, 393-404) whereby the latter has been chosen.
The problem which arose is that in the case of any thermostat and the thermo_modify, the pressure
related temperature (responsible for kinetic contributions) is automatically overwritten even when only
the temperature is modified. This effects the npt-iso calculation (using the temperature scalar) which
however appears seemingly correct in the output due to the thermo_modify alteration. The incorrectly
appearing pressures for npt-aniso/tri are actually correctly computed.
For a correct pressure it is necessary to create a compute of the default LAMMPS pressure
with the default temperature “thermo_temp” and specifiy it in both commands:
compute CSequ all temp/cs group_cores group_shells
compute thermo_press_lmp all pressure thermo_temp #correct kinetic scalar
(…)
thermo_modify temp CSequ press thermo_press_lmp
(and)
fix_modify thermostat_id temp CSequ press thermo_press_lmp
In the documentation and examples any core/shell pressure related information is missing
and I will shortly send an update as well as add another example for clarification. Based on
other mailing list requests I now browsed through, I clarified another issue of the core/shell
package usage and removed some confusing bits in the documentation.
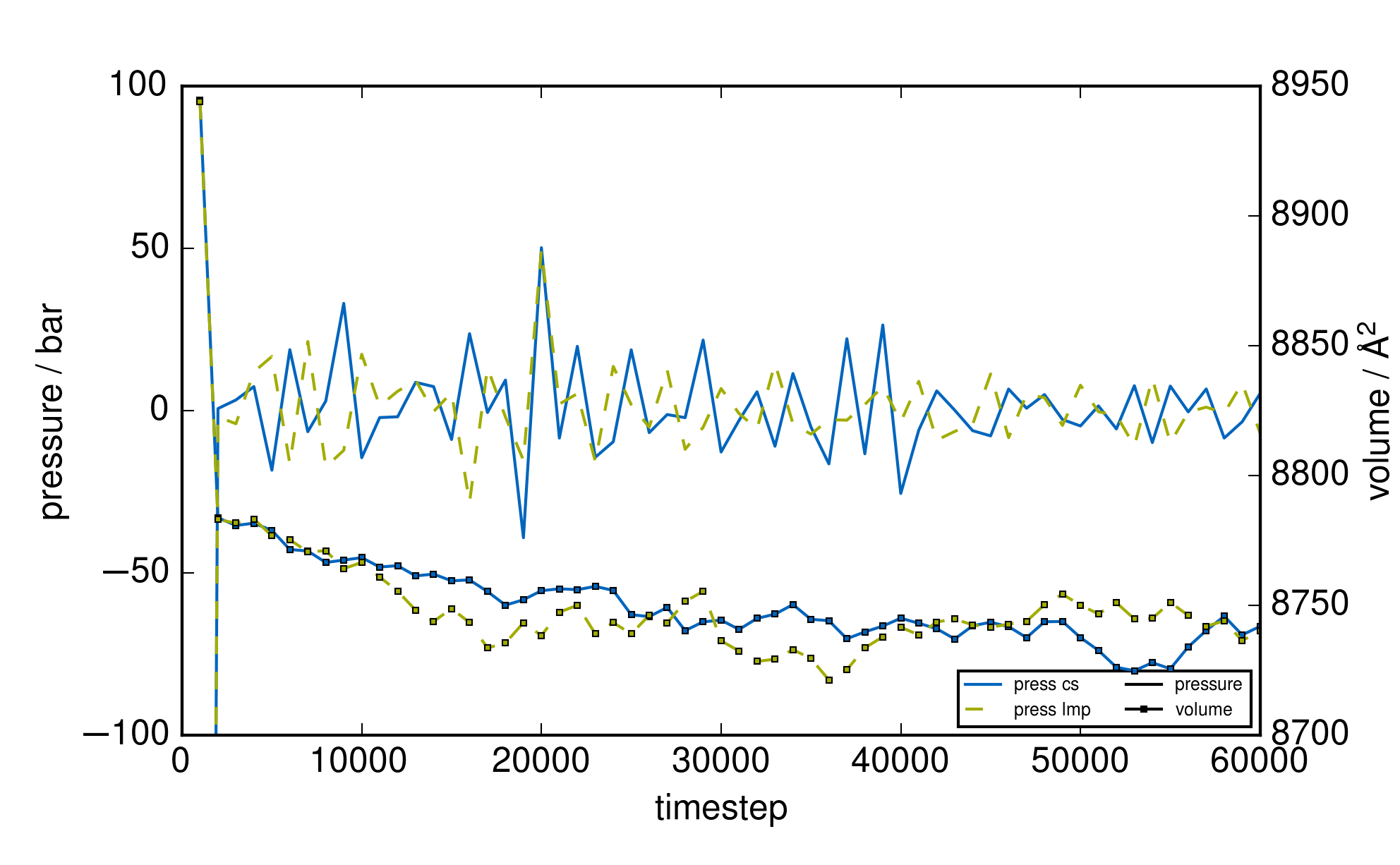
For the attached example, the new input leads to the correct pressure calculation and eleminates
all inconsistencies (see attached graph “plot_thermo_pressure_pto.png”). In general this error is
negligible and hardly noticable concerning pressure and volume in regular core/shell parametrizations
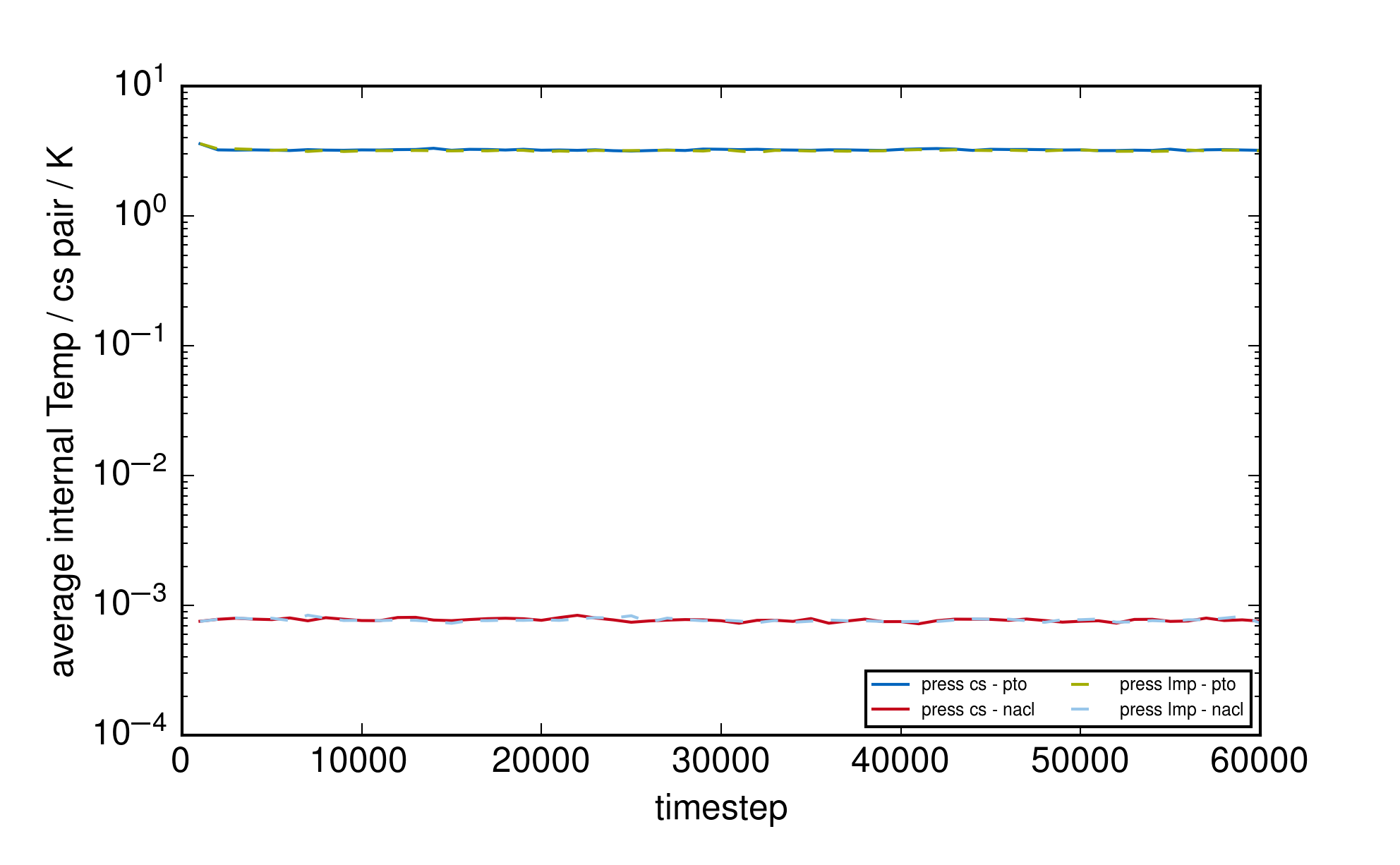
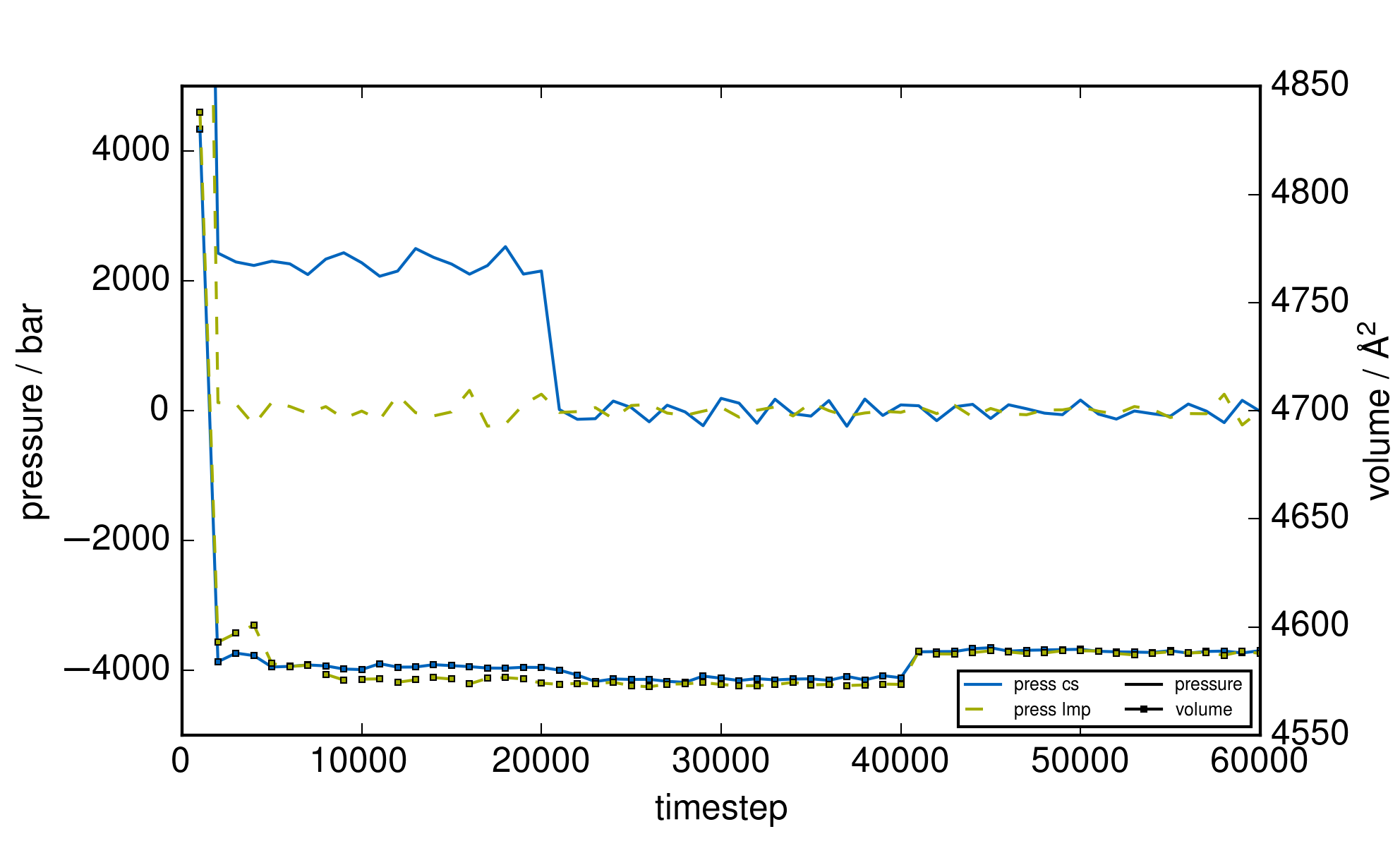
as the overall contribution of the core/shell oscillators to the kinetic energy is very small as seen for
nacl in the example directory (see attached graph “plot_thermo_pressure_nacl.png”).
In this context I would like to point out to be extremely cautious with the posted core/shell parametrization.
The spring constants of the core/shell bond are very high and I suspect them to be only stable due to
the anharmonic bond-potential and the small timestep used. This leads to a high internal energy of the
core/shell pairs (see attached graph “plot_cs_internal_temp.png” for comparison of the internal
temperatures of the posted example and the nacl-example). In the adiabatic core/shell model one
does not want high internal core/shell energies! They should be small and remain comparibly
constant as this is only an imaginary/non-physical degree of freedom.
Originally, the authors of this parametrization used the massless core/shell model (not implemented) where
internal kinetic energy is not an issue, but on the other hand the energy conservation of the simulation might
be affected, which they compensated with a very small timestep.
I hope this thread could be cleared up. Thanks again for pointing out the missing documentation.
Regards
Hendrik
PTO.data (28.4 KB)
PTO.in (2.66 KB)