Dear LAMMPS users,
I am using “pair_style ilp/graphene/hbn” for simulating 6 layers of Boron Nitride. Specific for this pair style, I think it is needed to specify each layer as a separate molecule. It is a bit challenging for me as I have a single data file that just contain atom coordinates, types and their charges. can I define them using another command separately? I can use the Z coordinate, as each layer has a unique Z distinguishable from the other one. How can I do that?
Best regards,
Runak
How do you generate the data file?
Since you need to set the molecule ID in the data file to have it available later, how did you assign it?
It would be easiest to define it there right away.
Other than that, since you have a layered material which is likely aligned with the coordinate system, you can just define a “block” region for one layer, create a group of atoms in that region and use the set command to set its molecule ID. Then delete region and group and start over with the next layer.
Thanks, It seems that it does not work as I am getting the error of too many neighbors which seems that have arise from bad configuration.
I have created the structure using ATOMSK software. It does not specify molecule ID to the particles, So I have define it manually. As the number of particles are huge, I have define a column of 1 where the molecule ID is expected and I want to redefine them in script properly. I think defining molecule ID is refine when simulation is running as particles move. Is there any other method. A command that can be running just at the beginning and does not repeat.
Best,
Three comments:
- if you are starting with a new and unfamiliar feature of LAMMPS, it is a very, very, VERY bad idea to do so with a very large system. You are just going to waste an enormous amount of computer time for no gain. Why not use a small test system to first figure out how to do things and then - once you get meaningful results - upgrade to a larger system.
- please pay better attention to what people suggest. I already gave you an outline of how to assign molecule IDs after the fact.
- please check the created geometry very carefully. That you are using an external tool does not guarantee it is correct (you could have provided bad instructions). You should particularly check for correct continuation across periodic boundaries and that appropriate box dimensions are used.
Thanks, I have done all of your mentioned tips. I changed the structure to a smaller one, set the correct molecule ID for each Boron Nitride layer and applied all of those tips in document of “pair_style ilp/graphene/hbn” including that the cutoff should be 16 and that pair style coul/shield should be used in combination of ilp/graphene/hbn. I have recently installed LAMMPS. Here is the potential format I have used:
pair_style hybrid/overlay tersoff ilp/graphene/hbn 16.0 coul/shield 16.0
pair_coeff * * tersoff BNC.tersoff B N
pair_coeff * * ilp/graphene/hbn BNCH.ILP B N
I have received the same error (too many neighbors, please check the configuration) but when I use this pair style, the program was running well.
pair_style hybrid lj/cut 8.0 tersoff
pair_coeff * * tersoff BNC.tersoff B N B N
pair_coeff 1 3 lj/cut 1.0 1.0
pair_coeff 1 4 lj/cut 1.0 1.0
pair_coeff 2 3 lj/cut 1.0 1.0
pair_coeff 2 4 lj/cut 1.0 1.0
Through this I have defined 4 types of B and N that belongs to different layers to be able define interlayer interactions.
I think there is a technical problem with “pair_style ilp/graphene/hbn” .
Best
… and I think there rather is a problem with how you are creating your geometries and data files.
As you see, everybody can easily make unsubstantiated claims, but there is a crucial component missing here: reproduciblility. You are making a lot of speculations but have no proof. That is very unscientific (just like my speculation that your initial geometry is bogus). Now, I have proof that the pair style is functioning as documented by pointing you to the example and confirming that it can complete without failure (I just reran it).
Now the burden of proof is on you to properly argue your case and provide convincing proof that the pair style is broken. Statements like “my input keeps producing the same error, no matter what I do” is not it unless you can provide the info and data (i.e. inputs) where others can check it. Please see the suggestions about what to do so that people will be more likely to look into your issues in the forum guidelines: Please Read This First: Guidelines and Suggestions for posting LAMMPS questions
Dear Axel,
Thank you for your attention.
I have attached the input file and the scripts. You would find two different scripts. One of them gave the error and other one has no problem.
I am using the final version of LAMMPS.
Best,
1.lmp (2.3 KB)
script2.txt (908 Bytes)
script.txt (804 Bytes)
Thanks, this just confirms that you and what you are doing are the main problem here. It also confirms that you are not paying attention to the forum guidelines as I have asked you to do. There are rather glaring issues here with your input and data files.
There is no such thing as a “final version” of LAMMPS. It would require that we would stop developing LAMMPS and we are far from it. It also is not a proper way to say the “latest”, which is likely what you meant (note, good science depends on accuracy and precision. what is the “latest” changes all the time and depends on whether you are using the feature release or a development version or a stable version). The way to specify the LAMMPS version is to give the exact version date. When I run LAMMPS, I get the following as the first line of output:
LAMMPS (2 Aug 2023 - Development - patch_2Aug2023-83-g9003e6ec77-modified)
which indicates, that I am using the “develop” branch from the git repository and a version that has 83 commits added (and some modifications of my own) since the release of the 2 Aug2023.
At the very least, you should have mentioned the version date, 2 Aug 2023. The latest would have been 15 June 2023 just a few days ago and the latest stable 23 June 2022 (but now those are both 2 August 2023).
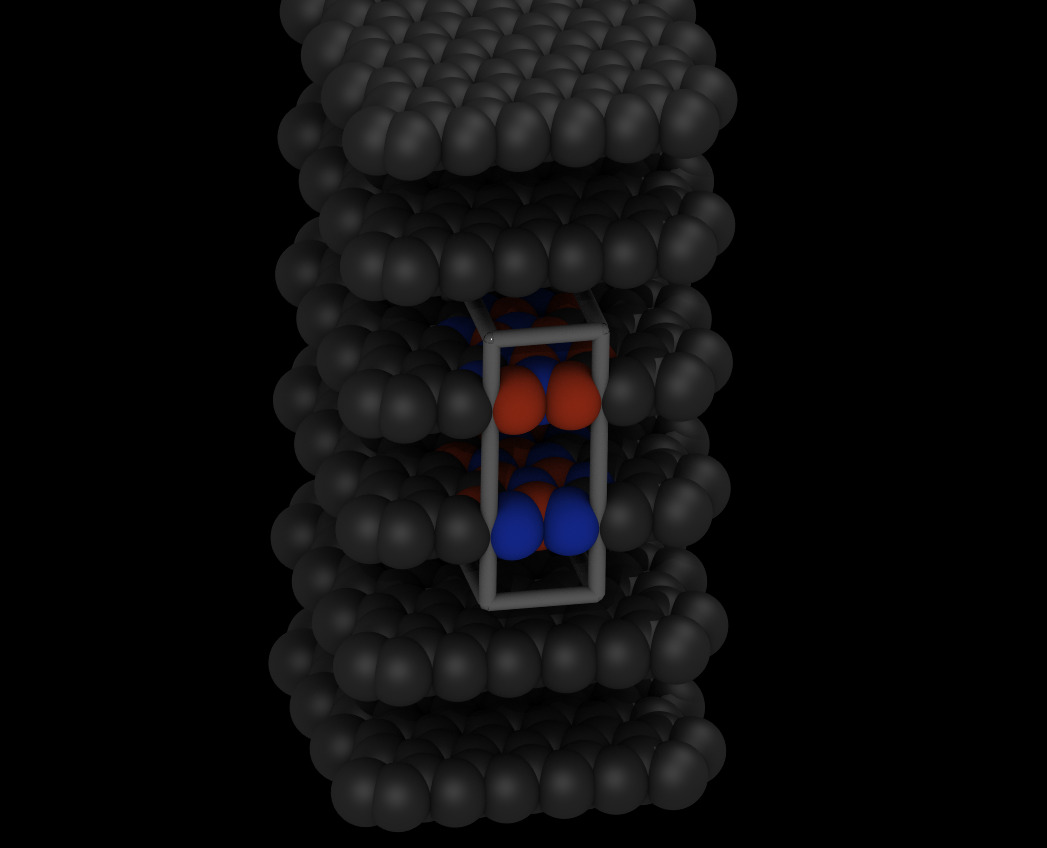
So I am looking at your data file. The first thing to do is to visualize it. That looks very odd:

The cell is far too small in one direction (up / down in this view) and too large orthogonal to it.
So if I turn on periodic images, it looks even worse:

Due to the too small box, atoms have rather close contacts.
I have empirically changed the box dimension, and that looks much better:
Now I am looking into your failing input file and it has some crazy stuff, too. For example:
region region1 block -200.0 212.56 -200.0 221.75 -100.0 207.70
create_box 4 region1
mass 1 10.81000000
mass 2 14.00700000
mass 3 10.81000000
mass 4 14.00700000
read_data input.lmp add append
Why on earth do you just a complex and convoluted thing? Why not just
read_data input.lmp
If your data file is correct, it will have the correct box dimensions, the correct number of atom types, the correct masses in addition to the atom positions. But now you are creating twice as many atom types as needed and the box dimensions are gigantic (over 400 angstroms in x and y and over 300 in z) while your data file has box dimensions smaller than 10 angstrom and even with the empirically adjusted version it is less than 15. You cannot get layers of sheets with that. As with the data file, a quick visualization shows how extremely crazy this setup is.

When following your trajectory to the point of where it stops with the “too many neighbors” error, you can see that the two “flakes” are bending and then the edges are touching, something that cannot happen with proper sheets, proper box dimensions, and proper periodic boundaries.

Indeed, with my empirically corrected box and the cleaned up and simplified input, it can run just fine and the sheets do not bend or touch. After replacing your fix nve command with
fix 1 all npt temp 300.0 300.0 1.0 aniso 1.0 1.0 10.0
It seems to nicely settle into some comfortable box dimensions and with flat not-ondulating sheets.

Overall, this is a nice demonstration of the GI-GO (i.e. garbage in, garbage out) principle and how applying proper checking, common sense and following best practices in setting up a simulation can create a proper simulation. This should give you some pause and you should spend some time on how you need to adjust your process and approach to setting up a simulation. Most certainly, you should approach the process with more humility and questioning yourself more than putting the blame on the software.
“I have empirically changed the box dimension, and that looks much better”
Dear Axel,
Thanks for your useful comments. I am sorry but I don’t know how you have changed the box of the input file. I think you have changed the input file but how? Have you changed the coordinates manually?
Is it possible to attach the modified files?
I have explained everything I did. You should be able to redo that. Some steps require some thinking, but that is a good thing, otherwise you would not learn anything.
Data files are text files and can be changed in a text editor just like input files.
I cannot upload files, because I have already deleted them. After confirming that my speculation (bad data file) was correct and confirming that the pair style can work as expected, there was no reason to keep those.