Hello,
I coded up my own version of Stillinger-Weber silicon using the default parameters in LAMMPS. At equilibrium my code produces the exact same value as LAMMPS; however, once I include dynamics there is a noticeable gap between my implementation and LAMMPS. I am not asking for anyone to debug my code rather it would be nice if someone could offer ideas as to what might be different.
The one thing I noticed is the LAMMPS docs specify the 3 body term to loop over i, i !=j, k > j whereas the SW paper denotes this sum as i > j > k.
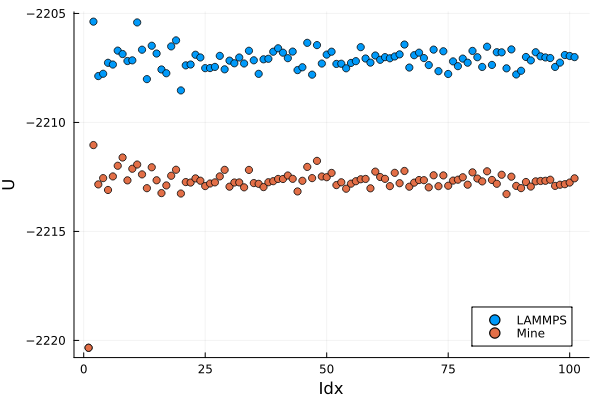
Here is the comparison, the x-axis is just an index into my array the actual spacing between points is 10ps.
If you get the same energy but different forces, then perhaps your force derivation is not the same.
Check out fix numdiff, perhaps it can help.
Also, @athomps may have some additional suggestions.
I’m just comparing energies at the moment, I have no derived the force calculation yet.
Forces are what matters in MD. Energies can have arbitrary offsets.
I agree, but wouldn’t you expect the offset to be identical at all time points? Here the offset fluctuates from ~4.75-5.5 eV.
I’m not exactly doing MD right now just using LAMMPS to double check my work.
Then I suggest you check each component of the energy/force computation individually.
The sw pair style is one of the oldest parts in LAMMPS, so if there would be a fundamental flaw in it, I would have expected that somebody in the last 20 or so years would have noticed.
How can you do “dynamics” without forces?
Sounds good Ill check agian.
I used LAMMPS to run dyanmics and printed the xyz and the energies. Then I used the positions to calculate the potential with my own implementation. I guess I just mean that its different at everything besides equilibrium.