I am trying to output the pairwise interaction energies in a system of TIP4P/2005 water (the pair_style is lj/cut/tip4p/long with pair_modify tail yes and kspace_style pppm/tip4p). It appears that the compute pair/local command does not work with this pair_style. Before I go into the source code to try to get the output, I was wondering if someone had tried to do implement this option and, if yes, what the main challenges were.
My plan was to go to the source code of compute pair/local and try to adapt that script to my needs.
Currently I use the 27. Oct 2021 version of LAMMPS.
you are using a long-range coulomb solver. there you can only output the real-space part as a pairwise contribution. the kspace part is an everything-with-everything contribution. if you want a decent approximation, you would have to record a trajectory and then using a cutoff coulomb interaction to have a pairwise decomposition where you also need to adjust the cutoff until you have a sufficiently converged result
the change you need to make is not with the compute pair/local command. It depends on the Pair::single() function of the pair style in question.
it is not possible to implement a consistent and correct Pair::single() function for a TIP4P pair style since that uses a different geometry for the coulomb interaction than for the LJ interaction. For that you need to know the identity and positions of all three atoms of the same water molecule, not just the distance between two atoms and their types and charges. You would have to ignore the provided distance argument. That is why no such function has been implemented for any existing TIP4P pair styles.
Could you please provide more details on how one would assess the convergence for this approximation to the k-space contribution of the Coulomb interactions, which you discuss in your first point? Do I vary the cutoff and track how the total energy of the particle changes when farther interactions are included?
In regard to your last point, do we not know the identity and positions of all three atoms at any given time? From my study of the source code, the organization of the data is such that always the molecules are organized by having first the O atom information, followed by the information for each of the H atoms.
I also was not explicit enough – I really only need pairwise interaction energies between water molecules. Because the LJ coefficients for H-H and O-H interactions are zero, the total pairwise interaction simplifies to the sum of the nine Coulomb energy contributions and the O-O LJ contribution. I may be missing something here, so I would appreciate any further guidance.
This should be self-evident. Please think about it for a bit.
The ordering of the atoms is indeed a requirement for the tip4p pair styles’ Pair::compute() function, but the Pair::single() function has different semantics. While you provide atom indices and types, you also provide the (squared) distance between those two atoms and expect to compute the force between (only!) those two atoms. However, for a tip4p pair style you have a different distance depending on whether you are considering the LJ interactions (where you consider the O atom location) and the Coulomb interactions (where you consider the M point location). In the latter case, you must project the forces back to the two H and O atoms. This can only be done if you ignore the provided distance and you cannot return the correct force since you are missing contributions from other pairs of the same molecule.
Thus if you want to correctly evaluate the pairwise contributions and forces, you would have to take your trajectory and post-process it to generate the missing M point locations and then you can use pair style lj/cut/coul/cut and compute all pairwise contributions for the four(!) sites of this water model as explained previously. The tip4p pair (and kspace) styles are an optimization to reduce the number of sites that need to be explicitly treated (and thus reduce the number of pairs to loop over substantially), but that will interfere with any kind of analysis in needs to include contributions from the M point.
I am trying to postprocess the trajectory as per your suggestion with the lj/cut/coul/cut. However, I run into a problem where I obtain too large (> 1000x) Coulombic interactions compared to the LJ interactions. As a simple example, for two atoms that are separated by distance r, I use
Elj = 4*epsilon*((sigma/r)^12 - (sigma/r)^6)
and
Ecoul = C*qi*qj/r
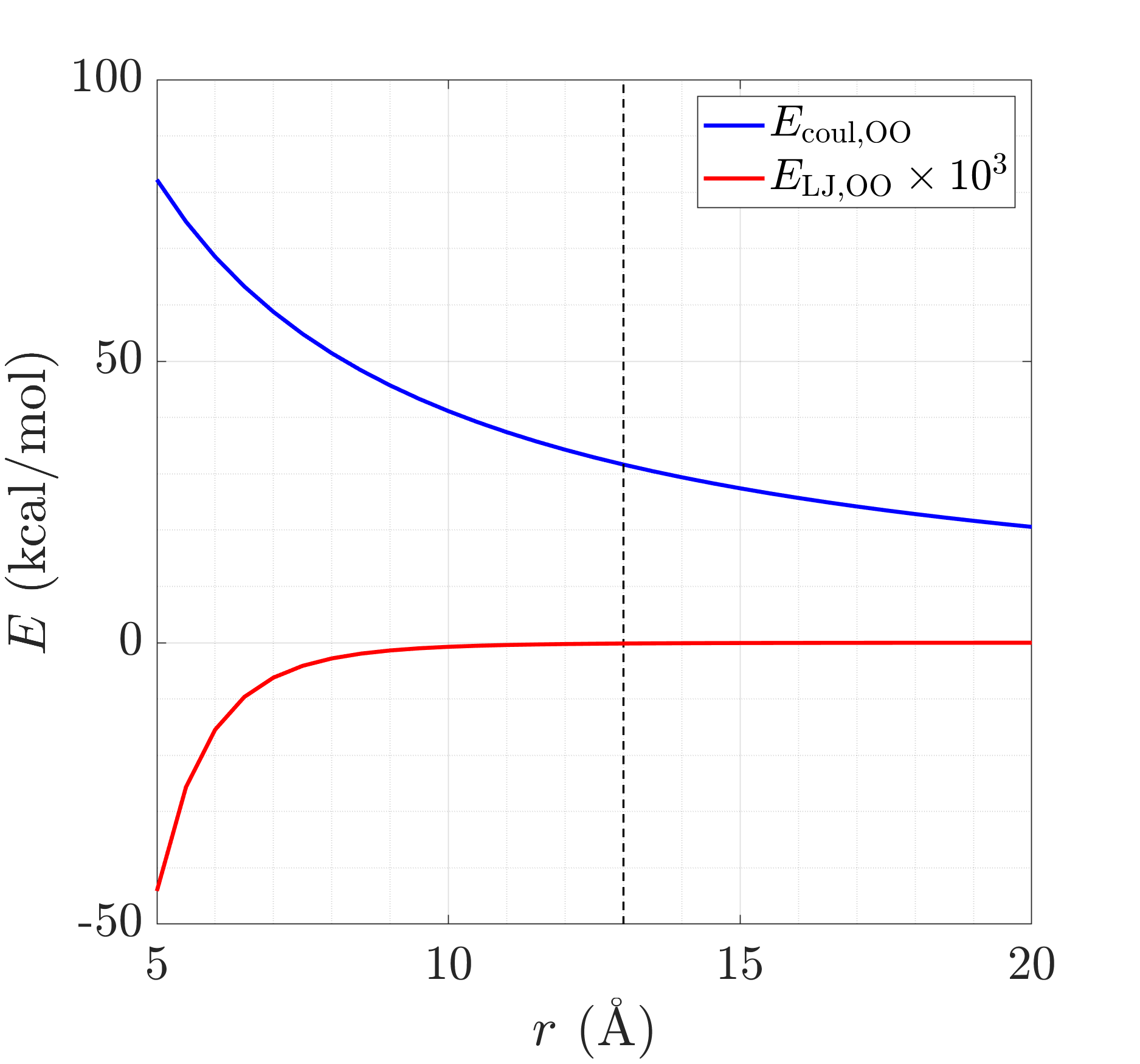
where C=332.06371 (from force.cpp), epsilon=0.1852, and sigma=3.1589 (for O atoms in TIP4P/2005 water). The contributions from the LJ and Coulombic part are plotted below. The black vertical line represents the cutoff that I use (Vega and de Miguel, J. Chem. Phys. 2007).
I think this problem arises because I incorrectly assume that factor_coul = 1. Could you please help me understand what the factor_coul variable represents and how it is calculated for lj/cut/coul/cut?
The factor_coul value is looked up from the special_bonds setting and it is indeed most commonly 1.0. If one or both atoms of the pair is involved in a bond that it value is changed to 0.0 or whatever is set by special_bonds depending on whether it is a 1-2, 1-3, or 1-4 pair. Usually, the same factor is applied for LJ interactions, so this is not the reason.
What is the distance for those close atoms? Which atoms are those? What are the assigned charges? This looks like you have been messing up something much more fundamental.
This makes no sense. When you are post-processing a TIP4P water trajectory where you reconstructed the point M position from H1, H2 and O (and thus have 4 atoms instead of 3 per molecule), then there should be NO pair of atoms for which you compute both LJ and Coulomb. TIP4P has LJ only between the O points, but those carry no charge and the M point and hydrogens have a charge but no LJ.