Here is a conversation I had with a LAMMPS user having trouble with
dihedral_style table. The issue appears to be resolved for now, but I
thought I'd forward it to the lammps list in case anybody else runs
into similar difficulty.
Andrew,
I wanted to let you know that I'm having problems trying to run my system in
parallel with the tabular dihedral potential. So I've done some analysis on
a trajectory where I spit out coordinates and data every timestep (which is
1 fs). When I run the system on 8 cores on my desktop after 1600 steps the
system blows up because some particles get too far apart (specifically what
happens is that I have a tabulated bond potential too and from one step to
the next a bond goes from equilibrium length to a length outside the table,
a 2 angstrom jump). When I run the system on 1 core this does not happen.
Going from 8 cores to 1 core the energies and pressures start deviating
significantly from one another after 250 or so timesteps. I'm unsure if
this is roundoff error or problems communicating between cores. I've
included a zip file that has all the data needed to run my system, it
includes a restart file, and input file and all the tabular potentials. Can
you let me know if your version of the program does the same thing? Thanks!!
----(files not included in forwarded email)---
I remember Axel's response to somebody complaining a few weeks ago
that there was a problem with
MPI. He said something to the effect that: "Problems in your setup
that were always there are more likely to show up when you run your
simulation in parallel." After looking at your files, I think that's
what happened.
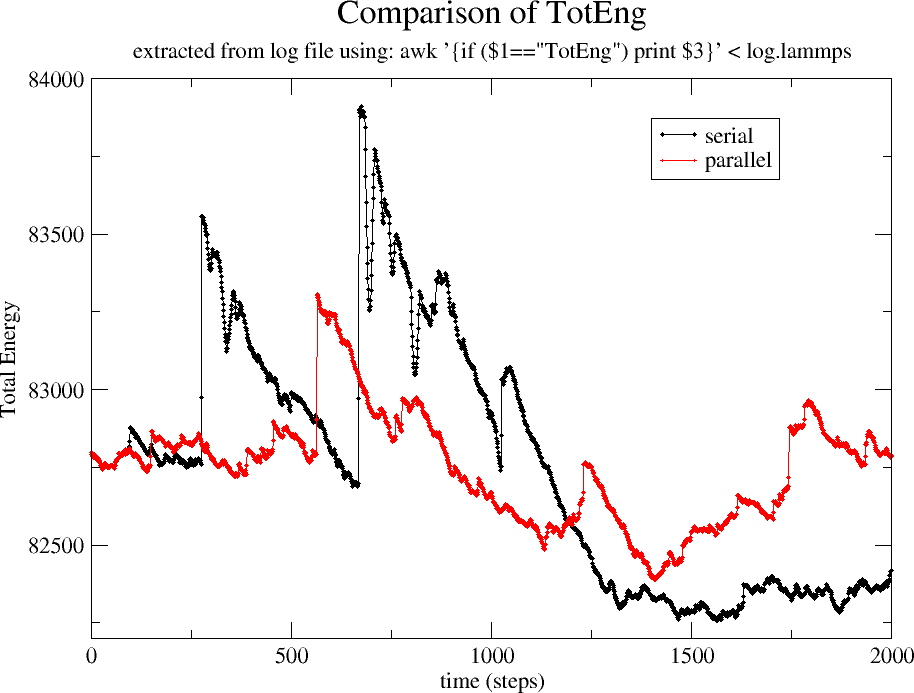
I see enormous (~500 kcal or kj) jumps in the total energy of your
system. These are a serious problem and they occur when running both
serial and parallel. (See attachment "compare_TotEng.png")
Coincidentally, the disagreement between the energies of the system in
serial and in parallel, start around the 100'th step, where the first
discontinuous jump in the energy occurs. Because the two simulations
diverge at exactly this moment, I also think that the problems you've
been having running in parallel are related to these energy
discontinuities.
--- As for why you have a problem with your energetics: ---
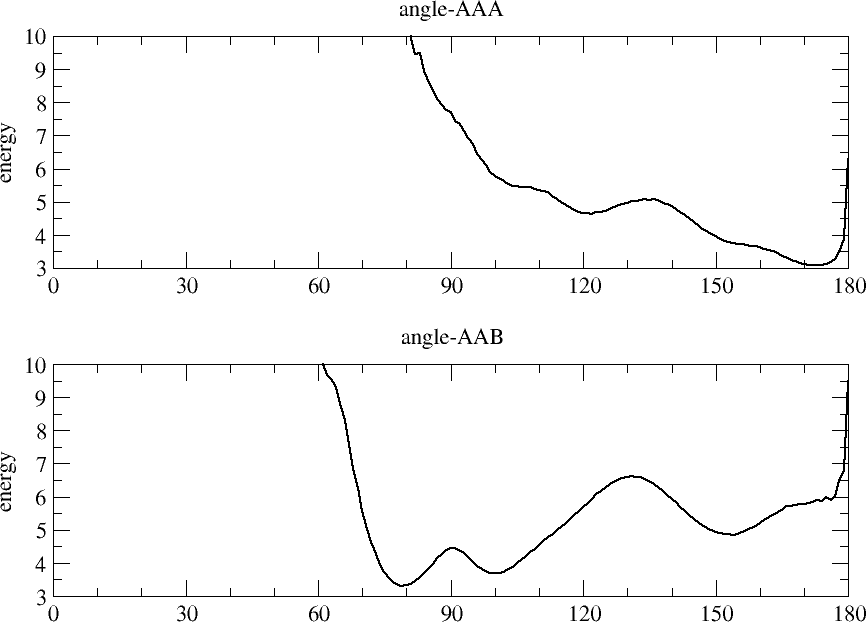
1) I think the most likely reason is that the angle-forces you are
using allow the polymer to adopt conformations with angles very close
to 180 degrees. (See "angle_tables.png".) There will always be a
singularity in any dihedral force field whenever 3 of the consecutive
atoms are colinear. Your current angle tables (AAA and AAB) allow the
angles to approach very close to 180.
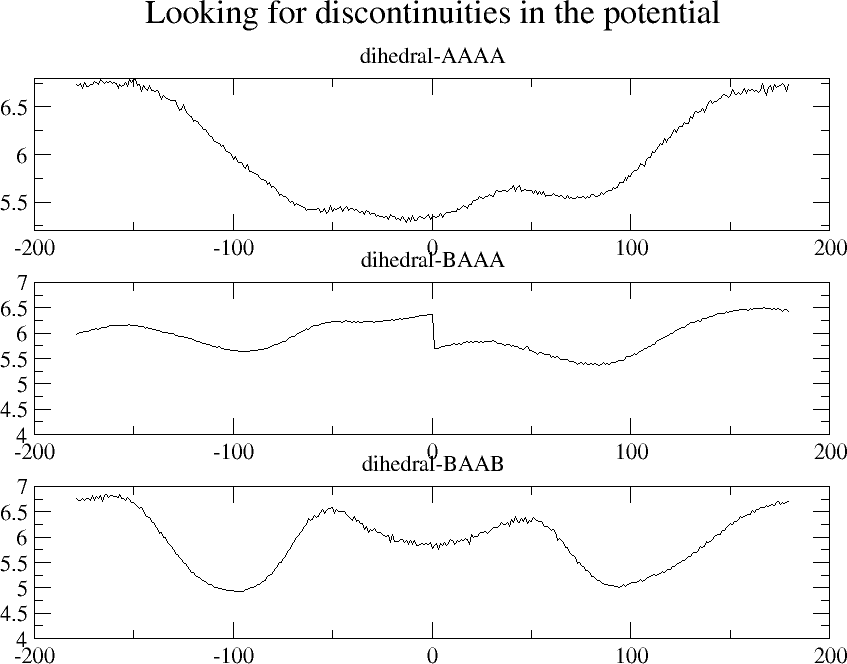
2) I also noticed a discontinuous jump in your BAAA dihedral table
(2nd graph in "dihedral_tables.png").