Hello everyone,
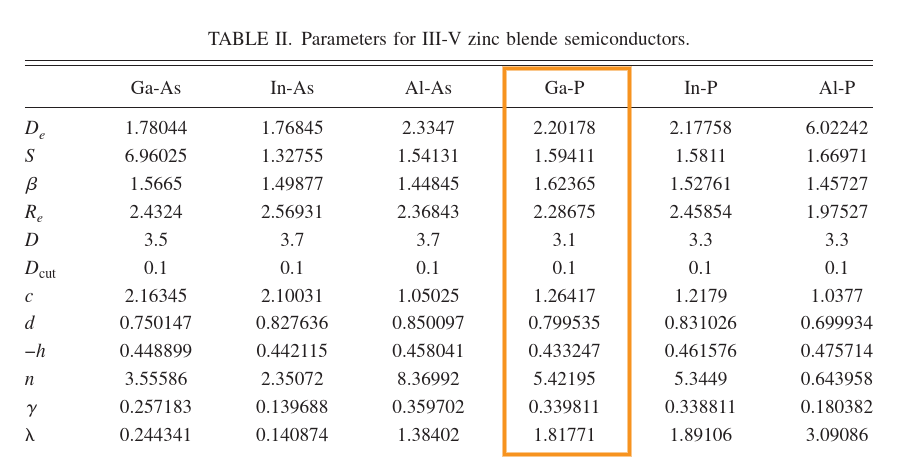
I am seeking advice regarding a molecular dynamics simulation in LAMMPS of Gallium Phosphide (GaP). I came across a paper by Powell et al.(Phys. Rev. B 75, 115202 (2007) - Optimized Tersoff potential parameters for tetrahedrally bonded III-V semiconductors) which parameterized a tersoff potential for Ga-P interaction, but it did not include the Ga-Ga and P-P interaction parameters. However, the author did find various elastic constants of GaP using that potential. On the other hand, I was able to successfully recreate the tersoff potential for GaN (included in the LAMMPS example directory) from a paper by Nord et al.(https://doi.org/10.1088/0953-8984/15/32/324), which included the Ga-Ga and N-N interaction parameters.
My question is, can I use the parameters from Powell et al. without including the Ga-Ga and P-P interaction parameters, or do I need to use some hybrid potential? I am not sure if this is possible or not. I would appreciate any guidance or suggestions on how to proceed with this issue.
Thank you.