Dear LAMMPS users,
Hi,
For parameters in Tersoff potential, we know that ( B, R, D, lambda1, A ) for C C C is ( 346.74 1.95 0.15 3.4879 1393.6 ), and for Si Si Si is ( 471.18 2.85 0.15 2.4799 1830.8 ) for SiC. But, I don’t understand how they end up with ( 395.1451 2.3573 0.1527 2.9839 1597.3111 ) for C Si Si. I also checked manual. But, It didn’t elaborate on it. Any comment please ?
Regards,
Ali
Dear LAMMPS users,
Hi,
For parameters in Tersoff potential, we know that ( B, R, D, lambda1, A )
for C C C is ( 346.74 1.95 0.15 3.4879 1393.6 ), and for Si Si Si
is ( 471.18 2.85 0.15 2.4799 1830.8 ) for SiC. But, I don't
understand how they end up with ( 395.1451 2.3573 0.1527 2.9839 1597.3111
) for C Si Si. I also checked manual. But, It didn't elaborate on it. Any
comment please ?
discussing parameters without also providing the units is a pointless exercise.
axel.
also, there is not *the* set of Tersoff parameters, but multiple
variants. the potential folder in LAMMPS has already 6 different files
with Tersoff parameters for C and Si.
please also note that information about those files is in the included
comments in the potential files directly in addition to what is
mentioned in the pair style documentation.
combined with the information in the referenced publications, you
should have every piece of information required.
axel.
Hi Axel,
Thank you very much for your responses. I checked that. But, there is a very big question on it. Have you noticed that the data you, I mean LAMMPS provider, used for SiC as Tersoff potential is based on the original Tersoff potential, which has Cosine format in fC term. However, pair style Tersoff has Sine format. I know the parameters are almost the same. But, Sine format is for a single component system, For instance pure Si, and for multi component system, such as SiC, we have to use Cosine format. for this situation I think you included Tersoff/table in LAMMPS. But, there is no input file example for using with Tersoff/Table and I believe that we have to include a new parameter 𝝌 in the input file. Is my understanding correct ?
Regards,
Ali
Hi Axel,
Thank you very much for your responses. I checked that. But, there is a very
big question on it. Have you noticed that the data you, I mean LAMMPS
provider, used for SiC as Tersoff potential is based on the original Tersoff
potential, which has Cosine format in fC term. However, pair style Tersoff
has Sine format. I know the parameters are almost the same. But, Sine format
is for a single component system, For instance pure Si, and for multi
component system, such as SiC, we have to use Cosine format. for this
situation I think you included Tersoff/table in LAMMPS. But, there is no
input file example for using with Tersoff/Table and I believe that we have
to include a new parameter 𝝌 in the input file. Is my understanding correct
?
your speculations are incorrect. tersoff/table is a user contribution
and was added primarily because it can run faster. it also differs
from the regular tersoff pair style in that it only supports one
variant of the tersoff potential, while the other supports 3, if i
remember correctly.
The Tersoff implementation in LAMMPS has been around for a very, very
long time. I was not involved in it, so i cannot make any
authoritative statement about it, but i cannot imagine that nobody
would have noticed a significant flaw in that long time. however, we
regularly have people posting claims that something is not correct on
the mailing list - especially like in your case with a lot of
unfounded speculation included - and most of the time, those were
proven wrong.
i am not claiming you are wrong, but you have to argue better and
provide much better evidence that your claim is correct, and the code
in LAMMPS is not doing what the documentation says it does.
don't expect people here to check things out for you. the burden of
proof is on your side.
axel.
I got your point on pair style: Tersoff/table. But, the input file shouldn’t be the same as pair style: Tersoff. I know that you included 6 different files for SiC. I checked the citation in them which are as follow:
Tersoff, Phys Rev B, 39, 5566 (1989)
Tersoff, Phys Rev Lett, 64, 1757 (1990)
Tersoff, Phys Rev B, 49, 16349 (1994)
The formula that they reference, and I attached an image from the formula section here, is different with the one that you have for pairstyle Tersoff. Have a look on equation ( 1c ) please. Moreover, look at 𝝌 in equation ( 1d ) which whouldn’t be 1 for SiC and it is 0.9776
Regards,
Ali
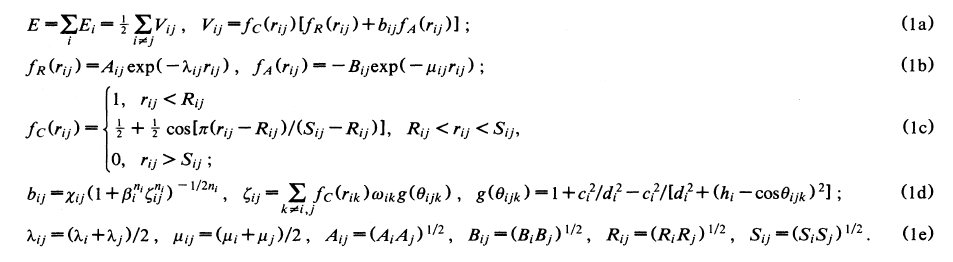
two (final) comments from me on this:
1) as i mentioned before, i am not an authority on this, so there is
no point in arguing with me and i will stop here. the name of the
person who authored a specific contribution is usually named in the
source code. if not, it is either a collaborative effort or was done
by one or more of the (at that time) core developers.
2) it doesn't matter what formula is displayed in the documentation;
it matters what is in the source code. it would be nice, if those are
identical, but this is a research software, and it is a fact of
research software, that sometimes the documentation is not fully in
sync with the source code. nobody does that intentionally, but it does
happen regularly, and not just for LAMMPS. thus unless you can provide
convincing proof, that the actual LAMMPS code does compute incorrect
results for a given correctly formatted parameter set and input deck,
there is little use in arguing.
axel.