I hope this email finds you well. I am writing to seek assistance regarding an issue encountered while simulating the interaction between coarse-grained polyurethane beads in LAMMPS.
To simulate the attractive interactions between the beads, I have assigned a strong mutual attraction for type 1 beads, a weak attraction between type 1 and type 2 beads, and also a weak attraction among type 2 beads. I have defined the cutoff range for the LJ potential as 2.50 and the length of the harmonic bond as 1.0.
To eliminate the influence of initial configurations, I have employed the “NVT” ensemble using the “fix nvt” command, with a target temperature of 1.0, a damping constant of 1.0, and a timestep of 0.001. Following this, I intend to proceed with further analysis using the “NPT” ensemble.
However, the simulation results indicate that many beads are in close proximity, and in some cases, they even overlap. The radial distribution function (RDF) plot exhibits significant peaks around 0, indicating pronounced aggregation between the particles.
I have attached the input and data files for your reference.
Additionally, I would like to provide information regarding my system setup. I am using Microsoft Windows Server 2019 Datacenter edition with an internal version number of 17763. The LAMMPS version I am using is LAMMPS (Large-scale Atomic/Molecular Massively Parallel Simulator) software, specifically the Git release/patch version from 27th October 2021.
I would greatly appreciate any guidance or insights you can provide to help resolve this issue. Thank you very much for your time and assistance.
There seems to be some problem in your RDF calculation, since (1) the LJ potential energy diverges as r → 0, so your run would crash long before particles can coincide; (2) the interparticle distance cannot be negative so what does “the RDF … around 0” even mean?
Your input script doesn’t show a compute rdf so you may have written your own and written it with bugs. As an additional method of analysis you should try to visualise your system so you can get an actual look at the configuration.
Thank you for your prompt response and clarification regarding the RDF calculation and the potential issues with my previous approach. I appreciate your insights on the matter.
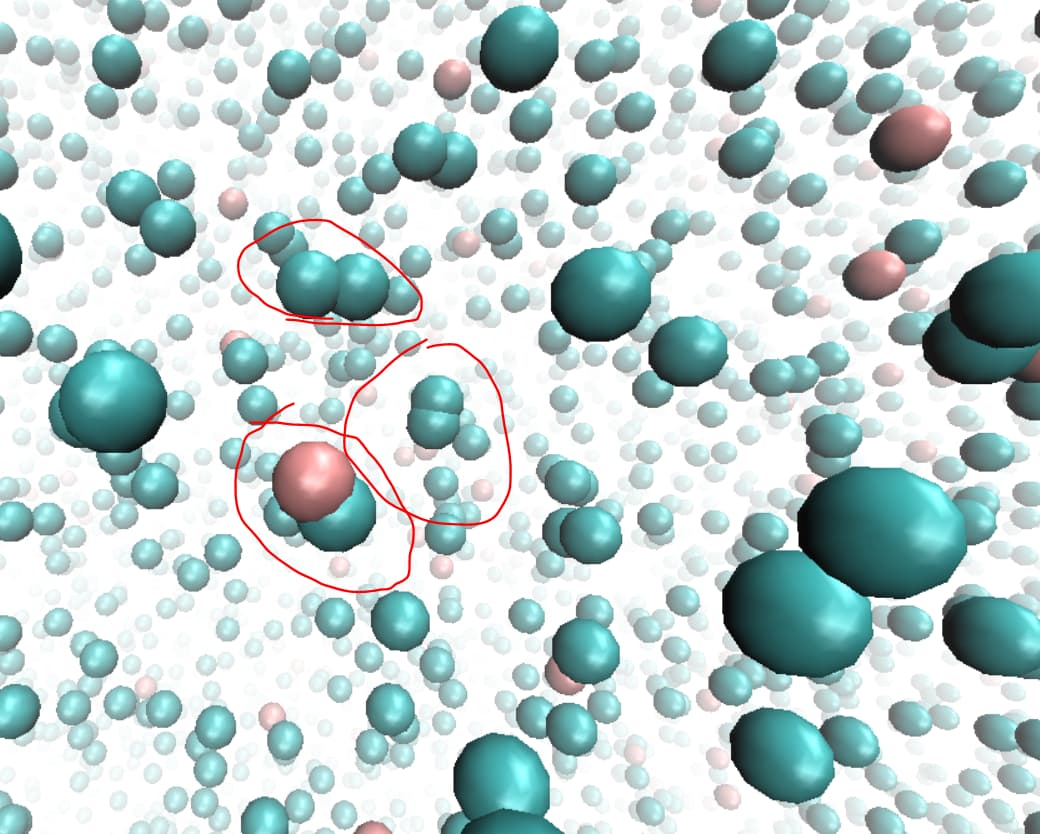
Following your suggestion, I have visualized the system using VMD to gain a better understanding of the atomic arrangement. I have attached a snapshot of the configuration at a specific moment for your reference.
Upon examining the visualization, I have indeed observed a significant degree of atomic overlap in a substantial portion of the system. This observation suggests that the interatomic distances between some atoms are indeed too close, potentially causing the observed overlap.
Considering your earlier points about the divergence of LJ potential energy as r approaches 0 and the impossibility of negative interparticle distances, it is clear that the concept of an RDF around 0 is not applicable, and my previous concern regarding the RDF calculation was misguided. I apologize for any confusion caused by my previous message.
To address the issue at hand, I will revisit the system setup and simulation parameters to ensure that the initial configuration and dynamics are correctly defined. Additionally, I will inspect the interatomic potential and interaction parameters to ensure their accuracy and suitability for the system under investigation.
Thank you once again for your valuable input and guidance. I will further investigate the matter and keep you updated on any progress or findings.