Hello,
I am trying to do a Particle hardening simulation where in a copper spot has been embedded in a Mo0.8075W0.0425Ta0.1Ti0.05Zr0.05 matrix. A sliced version of the triclinic simulation box is shown below (red circle is the copper spot). Eventually I plan to provide shear strain to this box to see if the dislocations “cut” or “bow through” the copper spot.
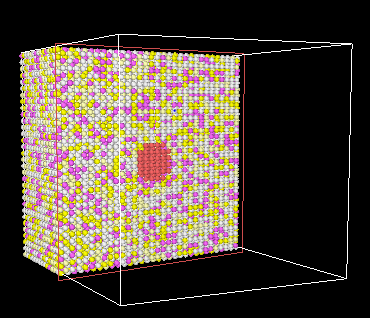
I am facing two problems in the program
-
The number of atoms in group Mo (Molybdenum) is becoming zero despite using any value in the type/fraction command of group Mo (type 2).
-
The pressure and other parameters are nan.
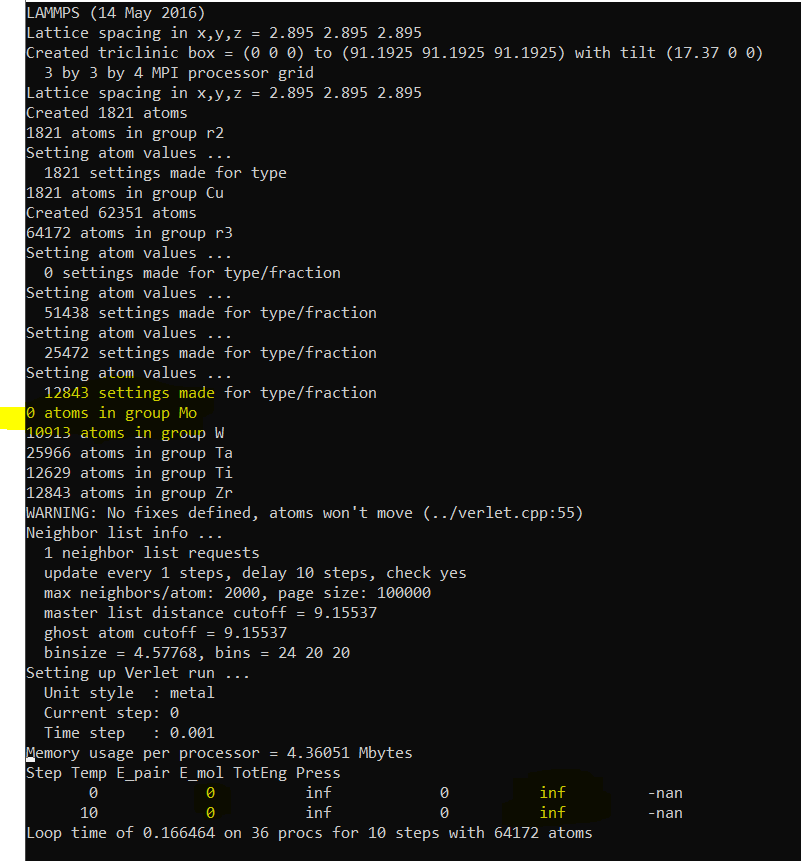
Also in the end I face this error
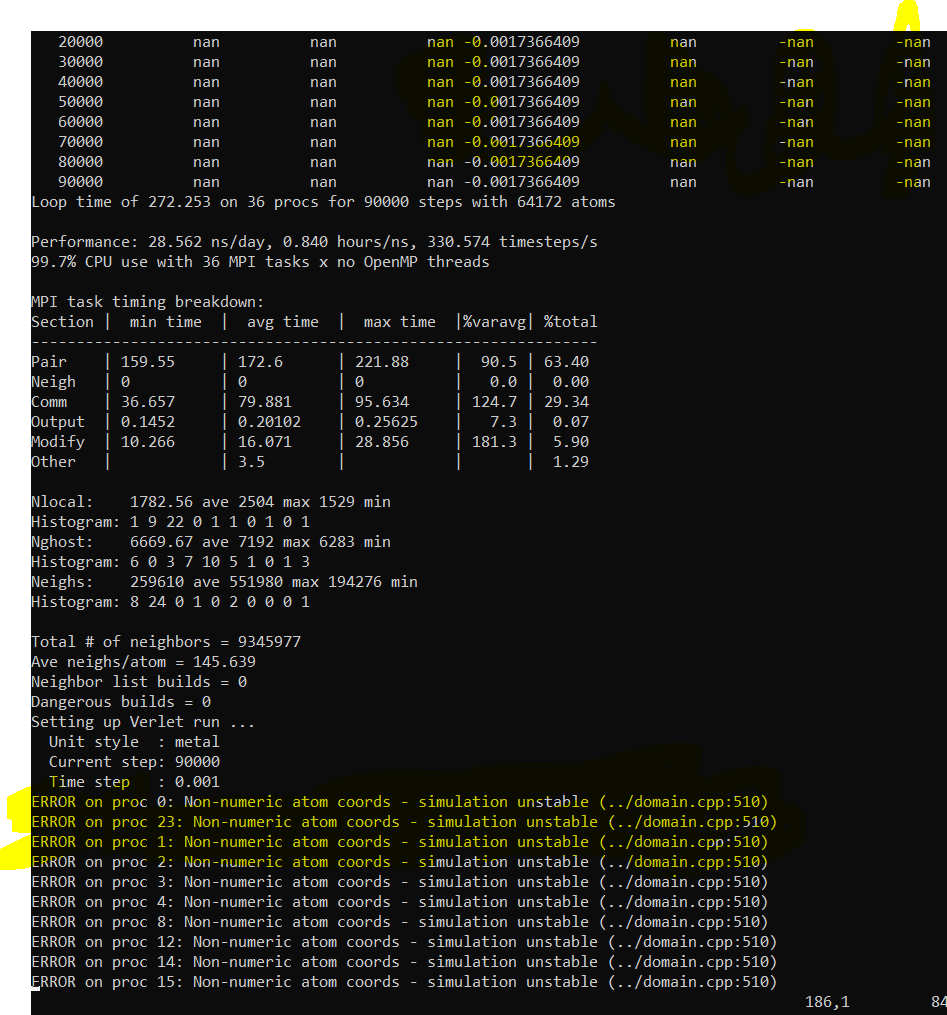
The code is here-
units metal
dimension 3
boundary p p p
atom_style atomic
variable latparam equal 2.895
----------------------- ATOM DEFINITION ----------------------------
lattice bcc ${latparam}
region whole prism 0 31.5 0 31.5 0 31.5 6 0 0
create_box 6 whole
lattice bcc ${latparam} orient x 1 0 0 orient y 0 1 0 orient z 0 0 1
region r2 sphere 15 15 15 6 side in
create_atoms 2 region r2
group r2 region r2
set group r2 type 1
group Cu type 1
region r3 sphere 5 5 5 3 side out
create_atoms 3 region r3
group r3 region r3
variable fa equal 14500
variable fb equal 12000
variable fc equal 5900
variable fd equal 2933
variable ft equal 62500
set type 2 type/fraction 3 (v_fa/v_ft) 1734536
set type 3 type/fraction 4 (v_fb/(14545)) 1734535
set type 4 type/fraction 5 (v_fc/(11955)) 1734534
set type 5 type/fraction 6 (v_fd/(5867)) 1734533
group Mo type 2
group W type 3
group Ta type 4
group Ti type 5
group Zr type 6
pair_style eam/alloy
pair_coeff * * MoTaTiWZrCu.set Mo Ta W Ti Zr Cu
dump 4 all custom 1000 dump.hea_equi id type x y z
run 10
print “-------Minimization SD------------------”
min_style sd
min_modify dmax 0.1
thermo 1000
thermo_style multi
minimize 1e-15 1e-15 100000 100000
reset_timestep 0
timestep 0.001
velocity all create 3000 123456 dist gaussian
fix 1 all npt temp 3000 3000 1 iso 0 0 1 drag 1
thermo 10000
thermo_style custom step temp press ke pe etotal vol density
0.09 nanosec run
run 90000
unfix 1
fix 2 all npt temp 3000 300 0.1 iso 0 0 1 drag 1
thermo 10000
thermo_style custom step temp press ke pe etotal vol density
0.09 nanosec run
run 1500000
unfix 2
fix 3 all npt temp 300 300 0.1 iso 0 0 1 drag 1
thermo 10000
thermo_style custom step temp press ke pe etotal vol density
0.09 nanosec run
run 90000
unfix 3
log log.quench_npt
fix 1 all npt temp 300 300 1 iso 0 0 1 drag 1
thermo 1000
thermo_style custom step temp press ke pe etotal vol density
2 nanosec run
run 10000
unfix 1
fix 2 all nvt temp 300 300 1
thermo 1000
thermo_style custom step temp press ke pe etotal vol density
run 20000
unfix 2
fix 3 all nve
thermo 1000
thermo_style custom step temp press ke pe etotal vol density
run 10000
unfix 3
log log.equi
dump 4 all custom 10000 dump.hea_equilibrate id type x y z
write_restart restart.tensile_equi
run 10000