Hello everyone,
I’m doing some lammps practice and I want create a perfect crystal for Fe (script flow the text)
But when I use OVITO to watch the dump file,
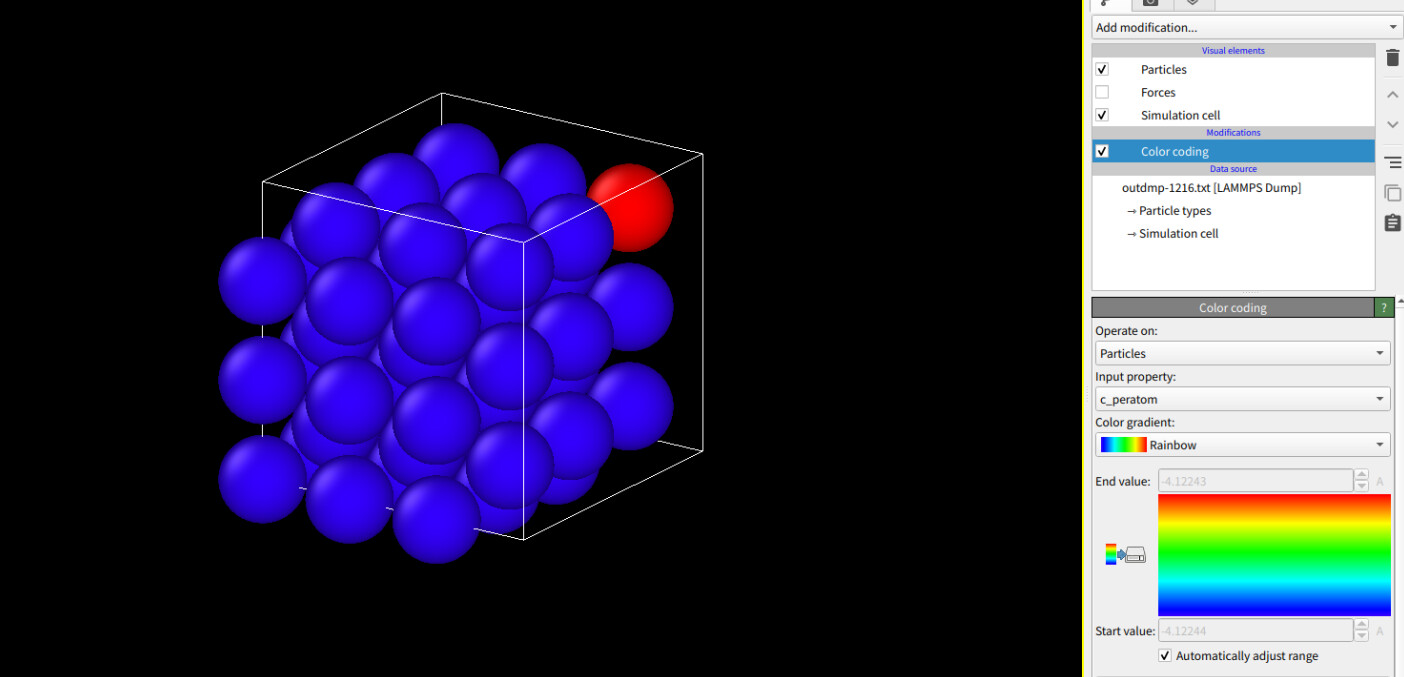
There is only one atom have the different values in potential energy.
If you can see the picture, the red one is that atom(values:-4.12243) and blue is all same( -4.12244).
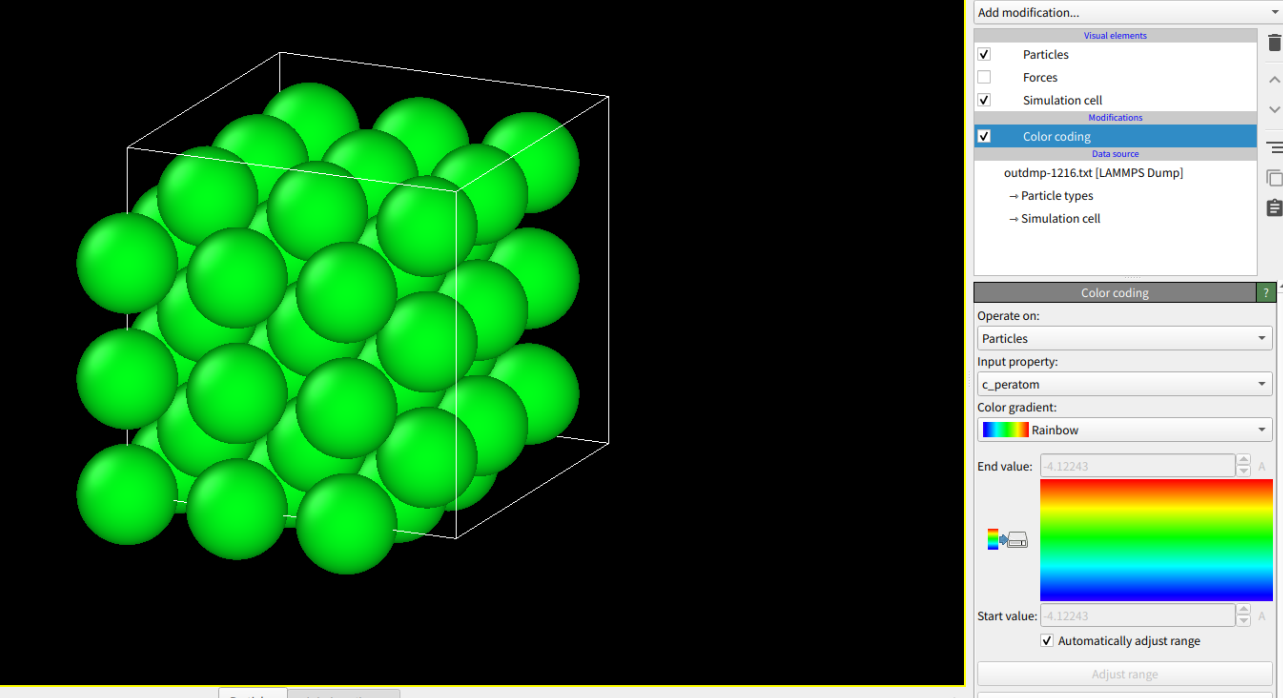
And when I change the ‘lattice bcc 2.8553’ to 2.855 or 2.8 or 2.6,
the result is all green(2.855: -4.12243; 2.8: -4.10052; 2.6: -3.6167)
Why is it like this? Did I miss something?
I am looking forward to your help. Many thanks.
There is my script:
variable x1 equal 3
variable x2 equal 3
variable x3 equal 3
units metal
boundary p p p
atom_style atomic
timestep 0.001
lattice bcc 2.8553
region box block 0 ${x1} 0 ${x2} 0 ${x3}
create_box 1 box
create_atoms 1 box
pair_style eam/fs
pair_coeff * * Fe_2.eam.fs Fe
neighbor 0.3 bin
neigh_modify delay 5
timestep 0.001
# ------------------------ compute pe,ke,stress ------------------------------
compute peratom all pe/atom
compute pe all reduce sum c_peratom
compute kinetic all ke/atom
compute ke all reduce sum c_kinetic
compute stress all stress/atom NULL
compute stress1 all reduce sum c_stress[1]
compute stress2 all reduce sum c_stress[2]
compute stress3 all reduce sum c_stress[3]
compute stress4 all reduce sum c_stress[4]
compute stress5 all reduce sum c_stress[5]
compute stress6 all reduce sum c_stress[6]
thermo 1000
thermo_style custom step lx ly lz press pxx pyy pzz pe c_pe ke c_ke c_stress1 c_stress2 c_stress3 c_stress4 c_stress5 c_stress6
# ------------------------ Create custom files with Von Mises Stress for Ovito viewing -----------------------------
variable stress_atom atom sqrt(((c_stress[1]-c_stress[2])^2+(c_stress[2]-c_stress[3])^2+(c_stress[3]-c_stress[1])^2+6*((c_stress[4])^2+(c_stress[5])^2+(c_stress[6])^2))/2)
variable etotal_atom atom c_peratom+c_kinetic
dump out all custom 1 outdmp-1216.txt id type x y z fx fy fz v_stress_atom v_etotal_atom c_peratom c_kinetic c_stress[2]
run 0