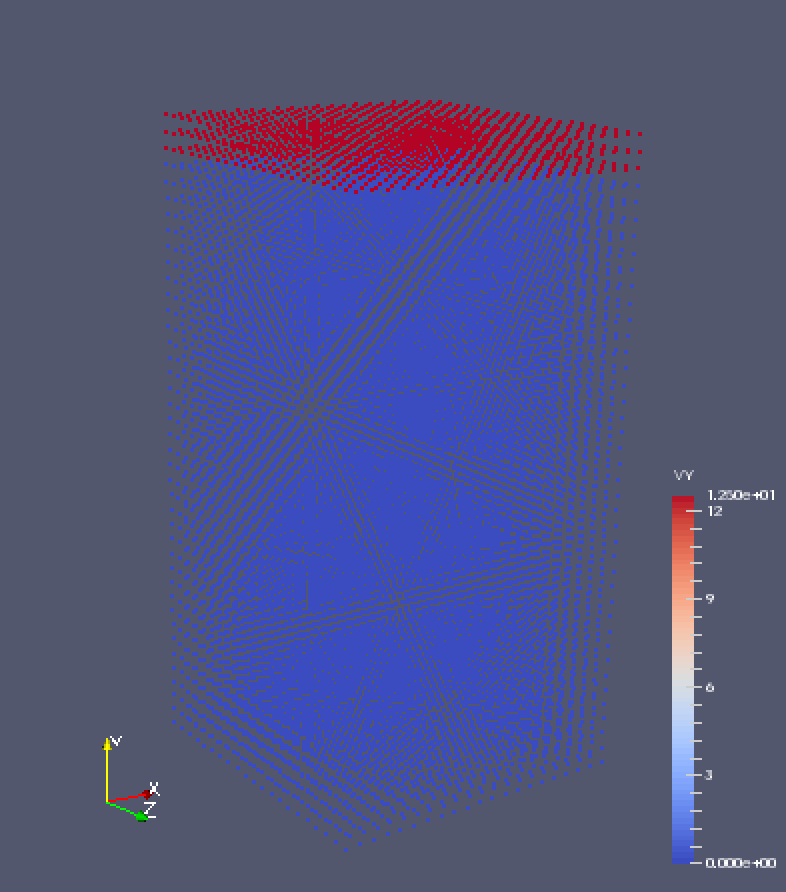
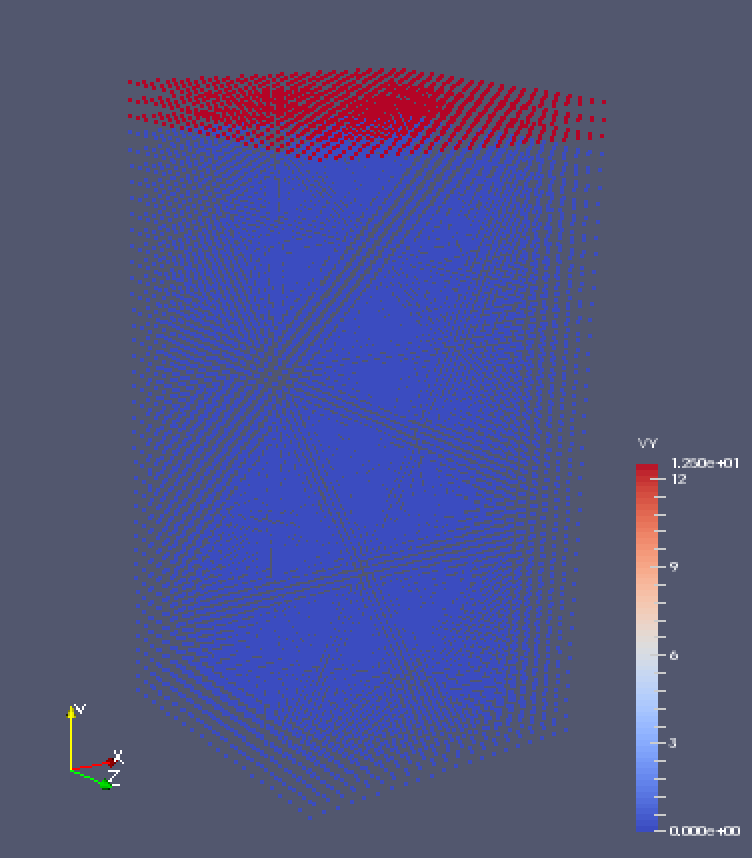
I am new to peridynamics and I have a problem with simulation of a tensile deformation of a block. The simulation box is 255025 (xyz), and the bottom is fixed. A constant y velocity is applied on the top layer. The script is attached.
The problem is the block does not deform at all though the velocity is properly observed when post-prosessing the results.
Two screen shots are attached. The first shows at step 0, the velocity on the top layer is 12.0. Then the second shows at the final step, the velocity remains the same. But the displacement is always 0. Also, the simulation box size in y direction remains unchanged, too. (box length in y direction is 50 at both step 0 and step final.)
units si
dimension 3
boundary s s s
atom_style peri
atom_modify map array
neighbor 3.75 bin
----------------------- ATOM DEFINITION ----------------------------
lattice sc 1.25
region whole block 0 25.0 0 50.0 0 25.0 units box
create_box 1 whole
create_atoms 1 region whole
------------------------ FORCE FIELDS ------------------------------
pair_style peri/lps
pair_coeff * * 45.0e3 15.0e3 3.75001 0.0005 0.25
set group all density 2200.0e-9
set group all volume 1.95
velocity all set 0.0 0.0 0.0 sum no units box
------------------------ Def Groups --------------------------------
region 1 block INF INF 47.5 INF INF INF units box
group uppergrip region 1
region 2 block INF INF INF 2.5 INF INF units box
group lowergrip region 2
group boundary union uppergrip lowergrip
group specimen subtract all boundary
------------------------ APPLY LOAD & BC ---------------------------
I am new to peridynamics and I have a problem with simulation of a tensile
deformation of a block. The simulation box is 25*50*25 (x*y*z), and the
bottom is fixed. A constant y velocity is applied on the top layer. The
script is attached.
The problem is the block does not deform at all though the velocity is
properly observed when post-prosessing the results.
Two screen shots are attached. The first shows at step 0, the velocity on
the top layer is 12.0. Then the second shows at the final step, the
velocity remains the same. But the displacement is always 0. Also, the
simulation box size in y direction remains unchanged, too. (box length in y
direction is 50 at both step 0 and step final.)
are the atoms that you assign the velocity to actually time integrated
(i.e. in the group for fix nve)? what you describe sounds as if they are
not.
I tried to replace “fix 1 specimen nve” in the script with “fix 1 all nve”, then I received an error message saying “Domain too large for neighbor bins”. Do you have any idea why do I receive this error message?
My guess is the material properties are not properly assigned that the whole sample is messed up. But I am not quite sure about this.
I tried to replace "fix 1 specimen nve" in the script with "fix 1 all
nve", then I received an error message saying "Domain too large for
neighbor bins". Do you have any idea why do I receive this error message?
My guess is the material properties are not properly assigned that the
whole sample is messed up. But I am not quite sure about this.
that would be my guess, too. probable the whole thing is exploding and
thus forcing a huge growth of the simulation cell. perhaps it is easier to
use fixed boundaries and leave some space.
there is a working examples in the LAMMPS distribution under examples/peri.
perhaps you should start with that and gradually modify it so that it
always works and you get to your desired system in stages and can see what
exactly goes wrong.