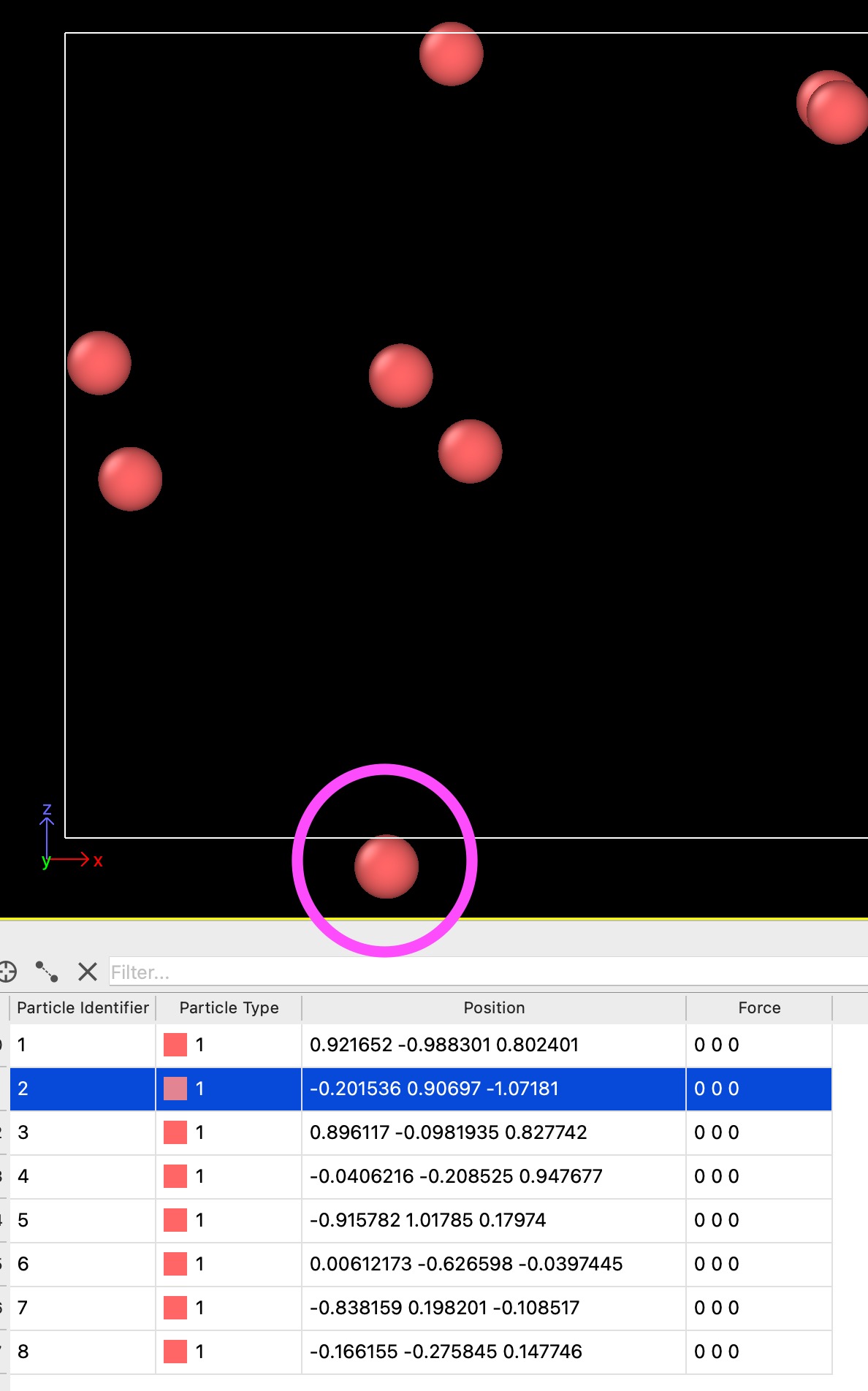
I just started using LAMMPS to perform some Brownian dynamics simulations. I am still working with simple noninteracting particles just to get used to the software. In one of my runs I noticed that the particles can get considerably outside the periodic box limits before being transferred to the opposite side. I am attaching a screenshot for clarity:
As you can see, particle 2 is visibly outside the box in the z-direction. This happens for ~25 timesteps before the particle is transferred to the top of the box, as it should. I believe that this is the intended behavior within LAMMPS but I could not find any info on how the periodic conditions are implemented or if it is possible to modify this behavior. Any insight would be greatly appreciated.
Periodic boundaries (well, actually non-periodic boundaries) in LAMMPS are realized as a special case of the domain decomposition. Rather than having only the principal set of atoms, LAMMPS manages “local” and “ghost” atoms, where ghost atoms are copies from the neighboring subdomains. With periodic boundaries those ghost atoms may be atoms from a domain that “wraps around”, i.e. is from the other side of the the simulation box. Since the positions of the ghost atoms are updated in every step, a strict enforcement of periodic boundaries is not required for as long as those atoms do not travel too far. The enforcement of periodic boundaries (and reconstruction of the ghost atoms) happens on every step where the neighbor lists are updated. If you output dumps only on steps where the neighbor lists are updated, atoms will always be inside the box. This all ties into the MPI parallelization of LAMMPS, more details are in the LAMMPS paper and in the LAMMPS documentation: 4.4. Parallel algorithms — LAMMPS documentation
If for some reason you need positions to be rewrapped into the the simulation box during output, there is the “pbc” keyword for that with the dump_modify command — LAMMPS documentation