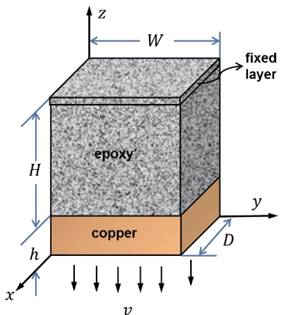
Hello,
I am a beginner of Lammps. Right now I want to do a tensile test with strain control method.
This is exactly what I want to simulate in Lammps.
To generate an interface and simulate pull-out test, I proceed with some steps
First, I generate an interface between epoxy and silica, and crosslinked epoxy resin under the 3D periodic boundary condition.
Second, I did 3 steps of equilibration simulation with crosslinked epoxy resin as the following input:
units real
atom_style full
boundary p p p
pair_style lj/cut/coul/cut 12.0 12.0
pair_modify mix arithmetic
dielectric 1.0
special_bonds lj 0.0 0.0 1.0 coul 0.0 0.0 1.0
bond_style harmonic
angle_style harmonic
dihedral_style charmm
improper_style harmonic
read_data 750-500_iter_7_step_5.data
# Equilibration Stage 2 (NPT dynamics at 600 K)
# Equilibration Stage 3 (NPT dynamics from 600 K --> 300 K)
# Equilibration Stage 4 (NPT dynamics at 300 K)
write_data data.*
Third, I need to change the boundary condition to the traction free boundary condition to simulate the tensile test. If I change boundary condition as f or s. The simulation aborted with an error message such as WARNING: Inconsistent image flags (…/domain.cpp:774) or ERROR on proc 0: Bond atoms 582 585 missing on proc 0 at step 0 (…/neigh_bond.cpp:65). How can I change boundary condition and fix the top of epoxy bulk ?
units real
atom_style full
boundary p p s
# Uniaxial Tensile Deformation
run 0
variable tmp equal "lz"
variable L0 equal ${tmp}
variable strain equal "(lz - v_L0)/v_L0"
variable p1 equal "v_strain"
variable p2 equal "-pxx/10000*1.01325"
variable p3 equal "-pyy/10000*1.01325"
variable p4 equal "-pzz/10000*1.01325"
variable p5 equal "lx"
variable p6 equal "ly"
variable p7 equal "lz"
variable p8 equal "temp"
variable t2 equal "epair"
variable t3 equal "ebond"
variable t4 equal "eangle"
variable t5 equal "edihed"
fix 1 all npt temp 300 300 50 x 0 0 1000 y 0 0 1000 drag 2
fix 2 all deform 1 z erate 1e-5 units box remap x
fix def1 all print 50 “{p1} {p2} {p3} {p4} {p5} {p6} {p7} {p8}” file epoxy_comp_100.def1.txt screen no
dump 1 all atom 50 500-200_iter_40_crosslink89-600-300-ps20-elastic.dump
dump_modify 1 scale no image yes
dump 2 all custom 50 500-200_iter_40_crosslink89-600-300-ps20-elastic.veldump vx vy vz
thermo_style custom step temp pxx pyy pzz lx ly lz epair ebond eangle edihed
thermo 50
timestep 1
reset_timestep 0
run 50000
unfix 1
unfix 2
unfix def1
unfix def2
Thank you and best regards,
Koochul