Dear LAMMPS users,
I have a problem which I cannot understand and therefore I cannot solve it. It would be very kind of you if you will explain me what I did wrong or give me some hint.
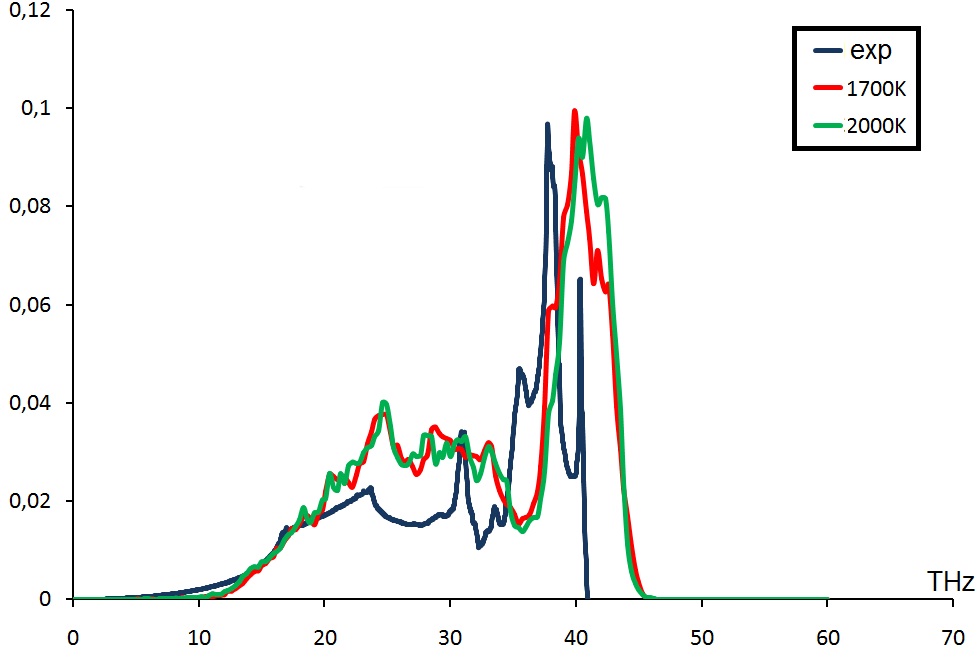
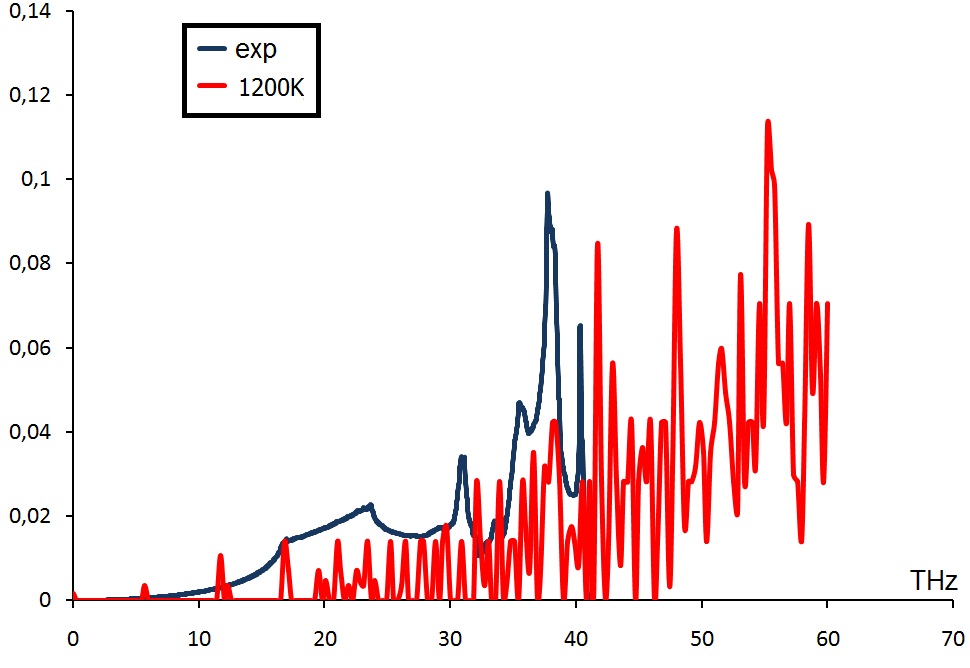
I want to calculate phonon DOS for diamond using LAMMPS fix command phonon with AIREBO potential. As I understand this type of calculation needs molecular dynamics simulation at finite temperature. My question is what temperature should I use to get a result comparable with experiment? Because I tried different temperatures from 300K to 1500K and results were not in an agreement with experimental data. Also I tried to use different sizes of diamond cell at fixed temperature but nothing changes. I tried to use ReaxFF potential as well, but it gets me even worse results.
Please find attached two pictures with phonon spectra obtained from AIREBO (AIREBO.jpg) and ReaxFF (ReaxFF.jpg).
Here is my input file for this calculation:
boundary p p p
units metal
atom_style atomic
read_data data.pos.new
pair_style airebo 3.0
pair_coeff * * CH.airebo C
neighbor 0.5 nsq
neigh_modify every 1 delay 0 check yes
fix 1 all npt temp 500 500 1 iso 1. 1. 1. pchain 8 drag 1.0
fix 2 all phonon 10 500 5000 map.in Phonon
timestep 2e-4
thermo_style custom step temp etotal press vol
thermo 100
dump 1 all xyz 500 out.md.xyz
dump_modify 1 append yes element C
run 650000
Thank you in advance!